

Unifying vertical and nonvertical evolution: a stochastic ARG-based framework
- PMID: 20525618
- PMCID: PMC2909786
- DOI: 10.1093/sysbio/syp076
Unifying vertical and nonvertical evolution: a stochastic ARG-based framework
Abstract
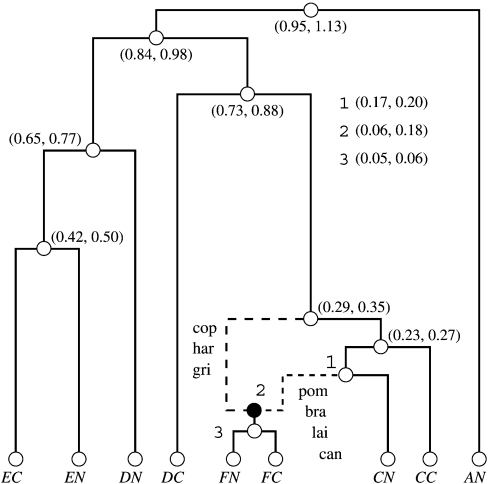
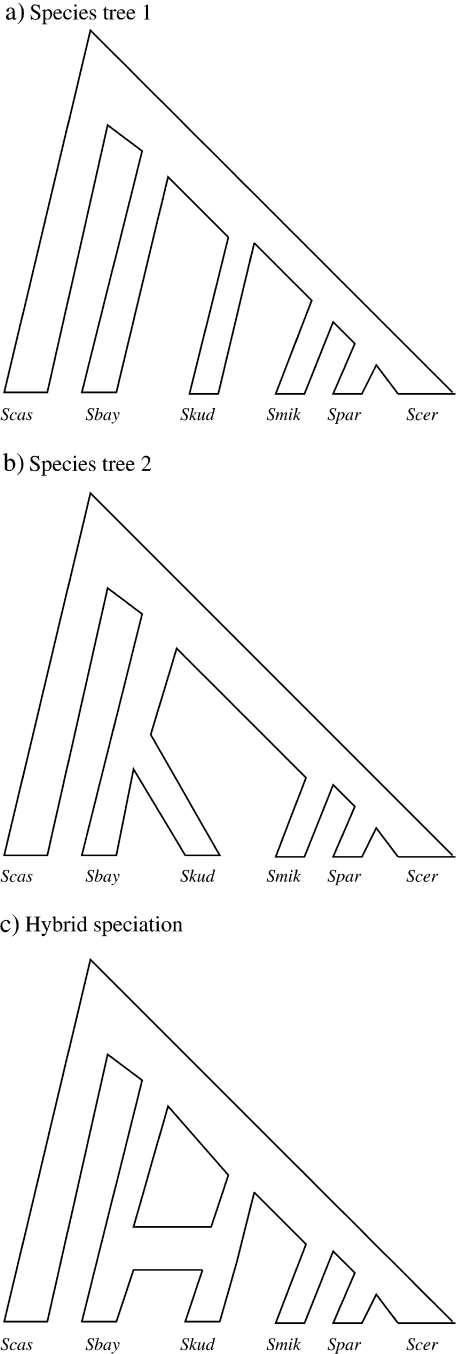
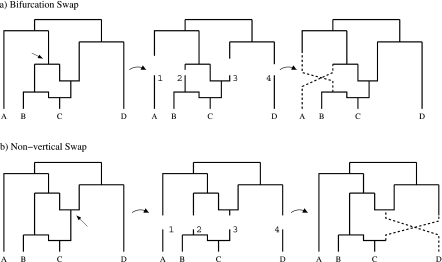
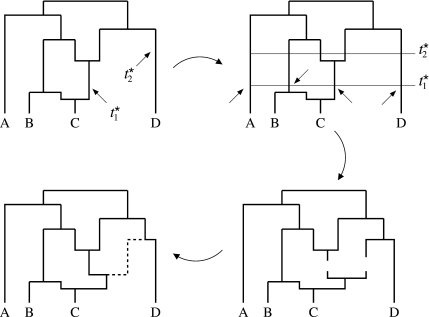
Evolutionary biologists have introduced numerous statistical approaches to explore nonvertical evolution, such as horizontal gene transfer, recombination, and genomic reassortment, through collections of Markov-dependent gene trees. These tree collections allow for inference of nonvertical evolution, but only indirectly, making findings difficult to interpret and models difficult to generalize. An alternative approach to explore nonvertical evolution relies on phylogenetic networks. These networks provide a framework to model nonvertical evolution but leave unanswered questions such as the statistical significance of specific nonvertical events. In this paper, we begin to correct the shortcomings of both approaches by introducing the "stochastic model for reassortment and transfer events" (SMARTIE) drawing upon ancestral recombination graphs (ARGs). ARGs are directed graphs that allow for formal probabilistic inference on vertical speciation events and nonvertical evolutionary events. We apply SMARTIE to phylogenetic data. Because of this, we can typically infer a single most probable ARG, avoiding coarse population dynamic summary statistics. In addition, a focus on phylogenetic data suggests novel probability distributions on ARGs. To make inference with our model, we develop a reversible jump Markov chain Monte Carlo sampler to approximate the posterior distribution of SMARTIE. Using the BEAST phylogenetic software as a foundation, the sampler employs a parallel computing approach that allows for inference on large-scale data sets. To demonstrate SMARTIE, we explore 2 separate phylogenetic applications, one involving pathogenic Leptospirochete and the other Saccharomyces.
Figures
References
-
- Altekar G, Dwarkadas S, Huelsenbeck J, Ronquist F. Parallel Metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics. 2004;20:407–415. - PubMed
-
- Amadal G. AFIPS Conference Proceedings. New York: ACM; 1967. Validity of the single processor approach to achieving large-scale computing capabilities; pp. 483–485.
-
- Andersson J, Sjögren A, Davis L, Embley T, Roger A. Phylogenetic analyses of diplomonad genes reveal frequent lateral gene transfers affecting eukaryotes. Curr. Biol. 2003;13:94–104. - PubMed
-
- Ané C, Larget B, Baum D, Smith S, Rokas A. Bayesian estimation of concordance among gene trees. Mol. Biol. Evol. 2007;24:412–426. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
