

Multi-Harmony: detecting functional specificity from sequence alignment
- PMID: 20525785
- PMCID: PMC2896201
- DOI: 10.1093/nar/gkq415
Multi-Harmony: detecting functional specificity from sequence alignment
Abstract
Many protein families contain sub-families with functional specialization, such as binding different ligands or being involved in different protein-protein interactions. A small number of amino acids generally determine functional specificity. The identification of these residues can aid the understanding of protein function and help finding targets for experimental analysis. Here, we present multi-Harmony, an interactive web sever for detecting sub-type-specific sites in proteins starting from a multiple sequence alignment. Combining our Sequence Harmony (SH) and multi-Relief (mR) methods in one web server allows simultaneous analysis and comparison of specificity residues; furthermore, both methods have been significantly improved and extended. SH has been extended to cope with more than two sub-groups. mR has been changed from a sampling implementation to a deterministic one, making it more consistent and user friendly. For both methods Z-scores are reported. The multi-Harmony web server produces a dynamic output page, which includes interactive connections to the Jalview and Jmol applets, thereby allowing interactive analysis of the results. Multi-Harmony is available at http://www.ibi.vu.nl/ programs/shmrwww.
Figures
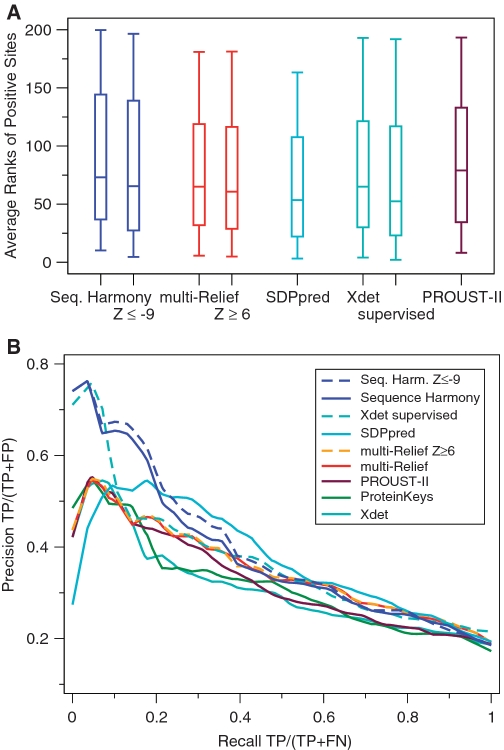
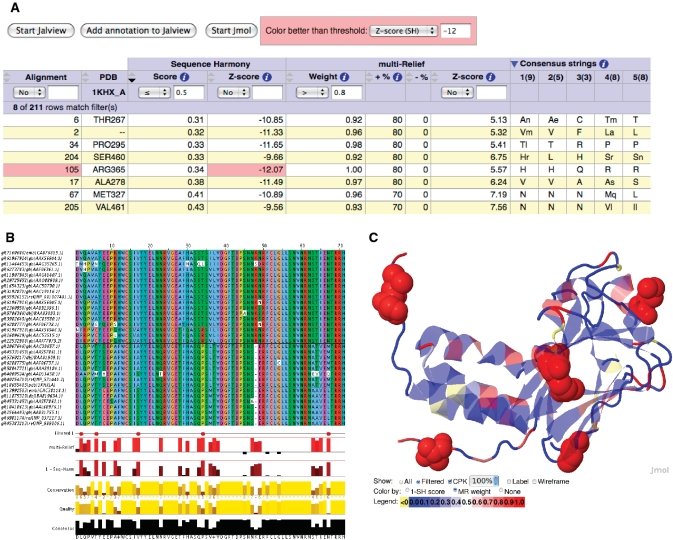
 0.5) and high mR weight (
0.5) and high mR weight ( 0.8). Only ALA278 at position 17 in the alignment is not a confirmed functional residue. The columns with arrows can be sorted. Most of these columns can also be filtered to display only those alignment positions that satisfy the user-supplied thresholds. (B) The output view in Jalview. Groups are outlined in the alignment and filtered positions (from the output table) are marked in the annotation track ‘Filtered 1’ with a tooltip detailing the filter like ‘Positions passing criteria [score
0.8). Only ALA278 at position 17 in the alignment is not a confirmed functional residue. The columns with arrows can be sorted. Most of these columns can also be filtered to display only those alignment positions that satisfy the user-supplied thresholds. (B) The output view in Jalview. Groups are outlined in the alignment and filtered positions (from the output table) are marked in the annotation track ‘Filtered 1’ with a tooltip detailing the filter like ‘Positions passing criteria [score  0.5; weight
0.5; weight  0.8] are indicated’. (C) View of the 3D context using Jmol with the protein coloured by mR weights, and filtered residues (from the output table) labelled and highlighted as space-filling spheres. Colouring by SH scores is also possible.
0.8] are indicated’. (C) View of the 3D context using Jmol with the protein coloured by mR weights, and filtered residues (from the output table) labelled and highlighted as space-filling spheres. Colouring by SH scores is also possible.Similar articles
-
Sequence harmony: detecting functional specificity from alignments.Nucleic Acids Res. 2007 Jul;35(Web Server issue):W495-8. doi: 10.1093/nar/gkm406. Epub 2007 Jun 21. Nucleic Acids Res. 2007. PMID: 17584793 Free PMC article.
-
Multi-RELIEF: a method to recognize specificity determining residues from multiple sequence alignments using a Machine-Learning approach for feature weighting.Bioinformatics. 2008 Jan 1;24(1):18-25. doi: 10.1093/bioinformatics/btm537. Epub 2007 Nov 17. Bioinformatics. 2008. PMID: 18024975
-
Sequence comparison by sequence harmony identifies subtype-specific functional sites.Nucleic Acids Res. 2006;34(22):6540-8. doi: 10.1093/nar/gkl901. Epub 2006 Nov 27. Nucleic Acids Res. 2006. PMID: 17130172 Free PMC article.
-
Jalview Version 2--a multiple sequence alignment editor and analysis workbench.Bioinformatics. 2009 May 1;25(9):1189-91. doi: 10.1093/bioinformatics/btp033. Epub 2009 Jan 16. Bioinformatics. 2009. PMID: 19151095 Free PMC article.
-
The PRALINE online server: optimising progressive multiple alignment on the web.Comput Biol Chem. 2003 Oct;27(4-5):511-9. doi: 10.1016/j.compbiolchem.2003.09.002. Comput Biol Chem. 2003. PMID: 14642759
Cited by
-
SigniSite: Identification of residue-level genotype-phenotype correlations in protein multiple sequence alignments.Nucleic Acids Res. 2013 Jul;41(Web Server issue):W286-91. doi: 10.1093/nar/gkt497. Epub 2013 Jun 12. Nucleic Acids Res. 2013. PMID: 23761454 Free PMC article.
-
Diversification in the HIV-1 Envelope Hyper-variable Domains V2, V4, and V5 and Higher Probability of Transmitted/Founder Envelope Glycosylation Favor the Development of Heterologous Neutralization Breadth.PLoS Pathog. 2016 Nov 16;12(11):e1005989. doi: 10.1371/journal.ppat.1005989. eCollection 2016 Nov. PLoS Pathog. 2016. PMID: 27851829 Free PMC article.
-
Both Intrinsic Substrate Preference and Network Context Contribute to Substrate Selection of Classical Tyrosine Phosphatases.J Biol Chem. 2017 Mar 24;292(12):4942-4952. doi: 10.1074/jbc.M116.757518. Epub 2017 Feb 3. J Biol Chem. 2017. PMID: 28159843 Free PMC article.
-
Experimental exchange of paralogous domains in the MLH family provides evidence of sub-functionalization after gene duplication.G3 (Bethesda). 2021 Jun 17;11(6):jkab111. doi: 10.1093/g3journal/jkab111. G3 (Bethesda). 2021. PMID: 33871573 Free PMC article.
-
The Rise and Fall of TRP-N, an Ancient Family of Mechanogated Ion Channels, in Metazoa.Genome Biol Evol. 2015 Jun 22;7(6):1713-27. doi: 10.1093/gbe/evv091. Genome Biol Evol. 2015. PMID: 26100409 Free PMC article.
