

A role for YY1 in repression of dominant negative LEF-1 expression in colon cancer
- PMID: 20525792
- PMCID: PMC2965227
- DOI: 10.1093/nar/gkq492
A role for YY1 in repression of dominant negative LEF-1 expression in colon cancer
Abstract
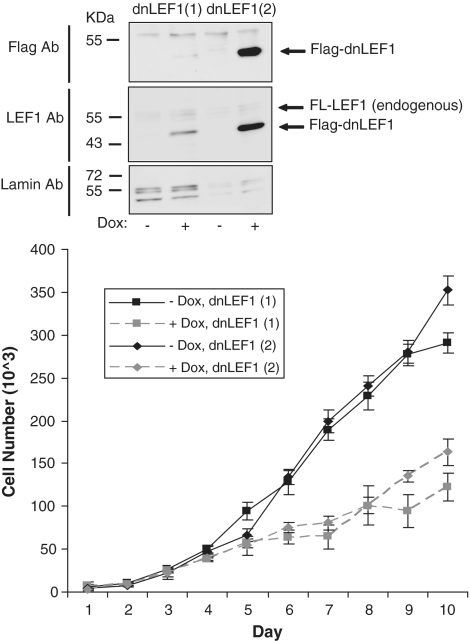
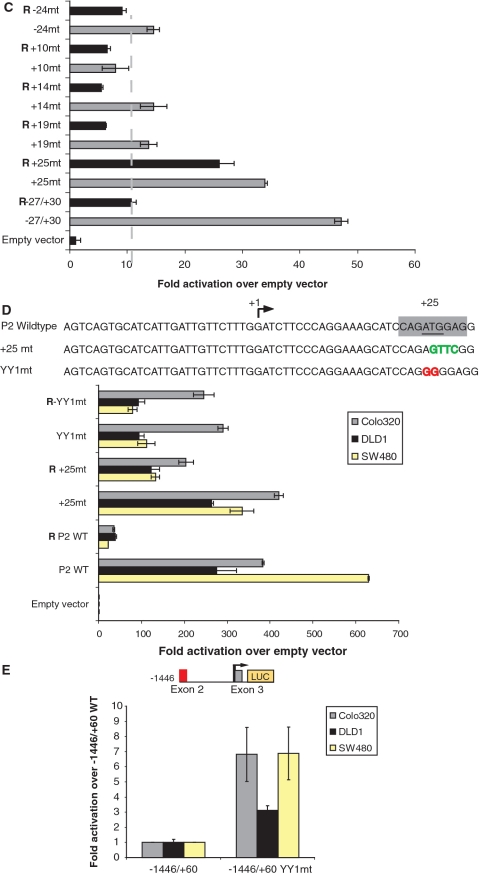
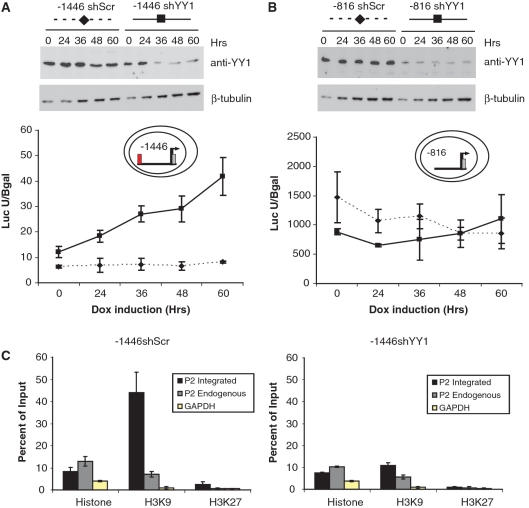
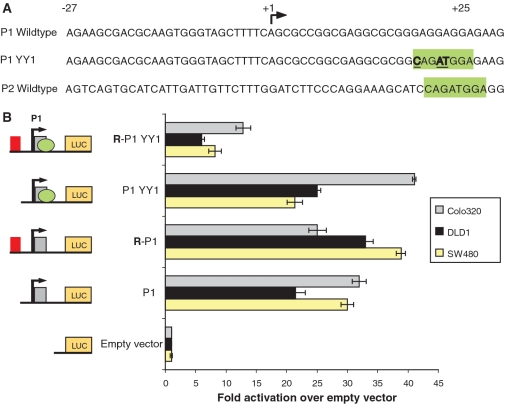
Lymphoid enhancer factor 1 (LEF-1) mediates Wnt signaling via recruitment of β-catenin to target genes. The LEF1 gene is aberrantly transcribed in colon cancers because promoter 1 (P1) is a Wnt target gene and is activated by TCF-β-catenin complexes. A second promoter in intron 2 (P2) produces dominant negative LEF-1 isoforms (dnLEF-1), but P2 is silent because it is repressed by an upstream distal repressor element. In this study we identify Yin Yang 1 (YY1) transcription factor as the P2-specific factor necessary for repression. Site-directed mutagenesis and EMSA were used to identify a YY1-binding site at +25 in P2, and chromatin immunoprecipitation assays detected YY1 binding to endogenous LEF1 P2. Mutation of this site relieves P2 repression in transient transfections, and knockdown of endogenous YY1 relieves repression of integrated P2 reporter constructs and decreases the H3K9me3 epigenetic marks. YY1 is responsible for repressor specificity because introduction of a single YY1-binding site into the P1 promoter makes it sensitive to the distal repressor. We also show that induced expression of dnLEF-1 in colon cancer cells slows their rate of proliferation. We propose that YY1 plays an important role in preventing dnLEF-1 expression and growth inhibition in colon cancer.
Figures



References
-
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1784–1787. - PubMed
-
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. - PubMed
-
- Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science. 1997;275:1790–1792. - PubMed
-
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. - PubMed
