

The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing
- PMID: 20526329
- PMCID: PMC2919364
- DOI: 10.1038/ncb2070
The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing
Erratum in
- Nat Cell Biol. 2010 Dec;12(12):1249
Abstract
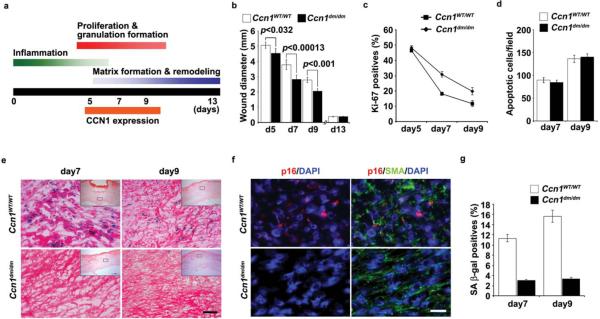
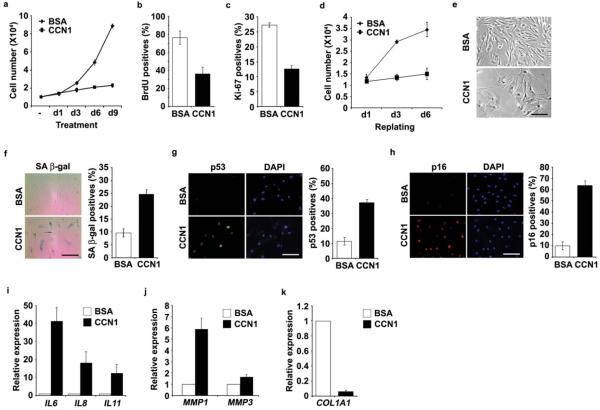
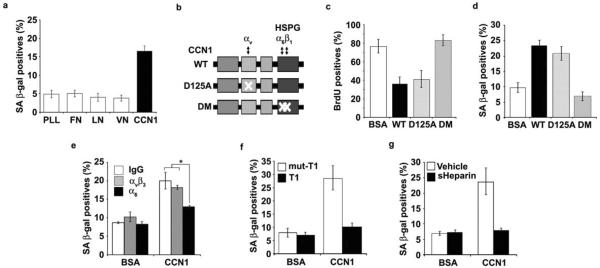
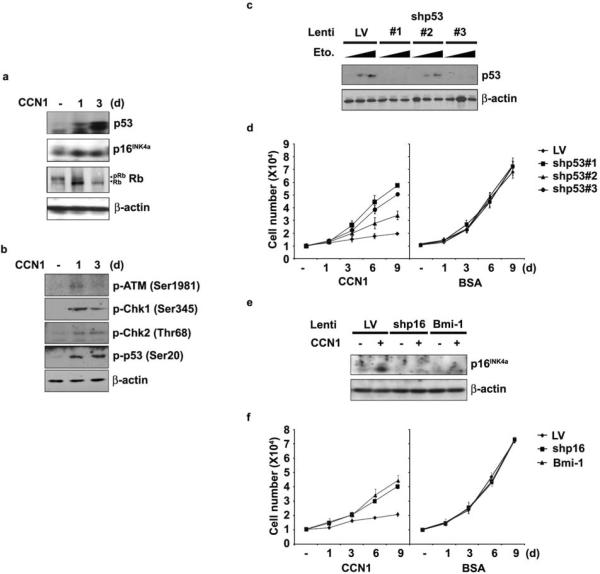
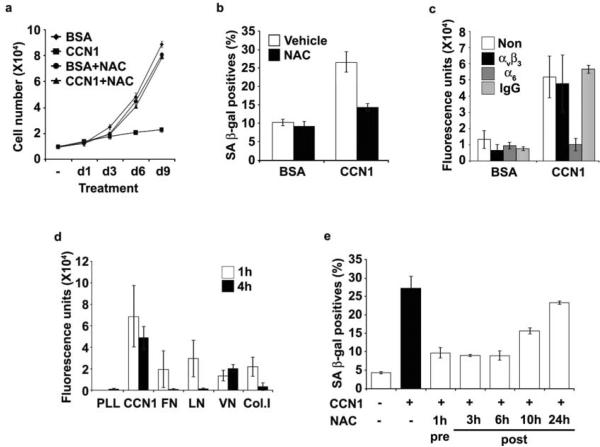
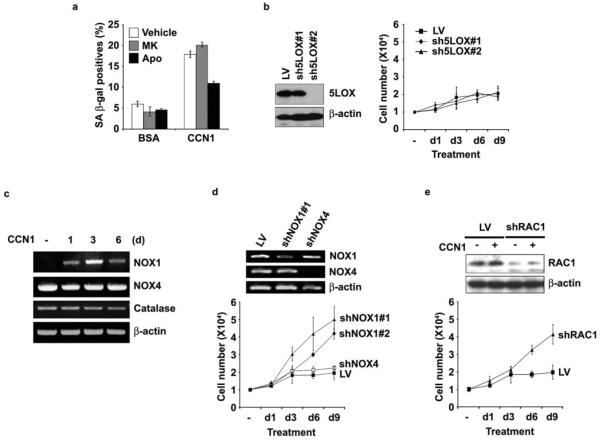
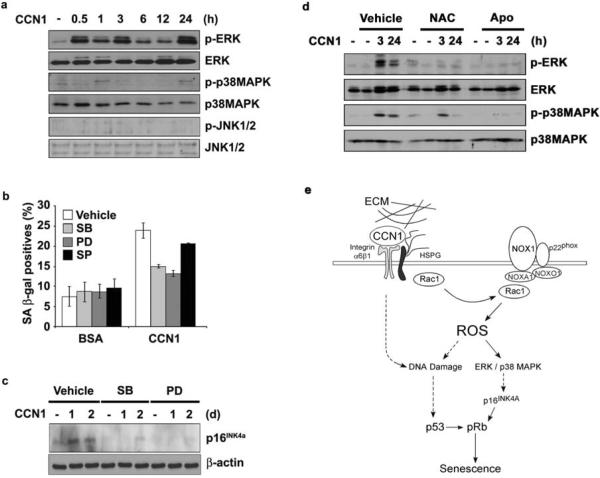
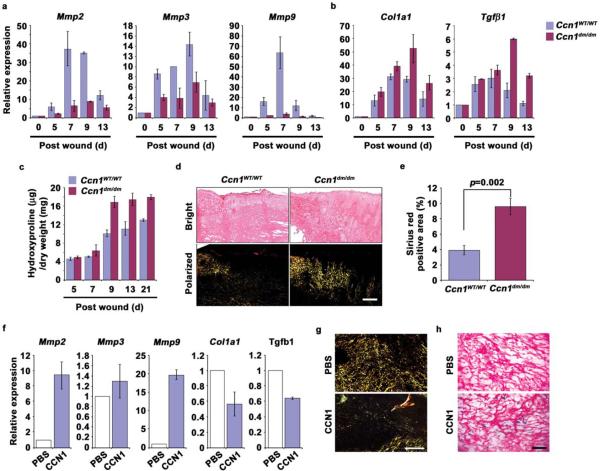
Cellular senescence is a recognized mechanism of tumour suppression; however, its contribution to other pathologies is not well understood. We show that the matricellular protein CCN1 (also known as CYR61; cysteine-rich protein 61), which is dynamically expressed at sites of wound repair, can induce fibroblast senescence by binding to integrin alpha(6)beta(1) and the heparan sulphate proteoglycans (receptors involved in cell adhesion). CCN1 induces DNA damage response pathways and activates p53 and the RAC1-NOX1 complex, which generates reactive oxygen species (ROS). This results in the ROS-dependent activation of the p16(INK4a)/pRb pathway, leading to senescence and concomitant expression of antifibrotic genes. Senescent fibroblasts accumulate in granulation tissues of healing cutaneous wounds and express antifibrotic genes in wild-type mice. These processes are lost in knockin mice that express a senescence-defective Ccn1 mutant, resulting in exacerbated fibrosis. Topical application of CCN1 protein to wounds reverses these defects. Thus, fibroblast senescence is a CCN1-dependent wound healing response in cutaneous injury that functions to curb fibrosis during tissue repair.
Figures
References
-
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965;37:614–636. - PubMed
-
- Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007;8:729–740. - PubMed
-
- Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. - PubMed
-
- Braig M, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
