

doi: 10.1186/1710-1492-1-4-142.
Epub 2005 Dec 15.
Pediatric hemophagocytic syndromes: a diagnostic and therapeutic challenge
Affiliations
- PMID: 20529219
- PMCID: PMC2877072
- DOI: 10.1186/1710-1492-1-4-142
Item in Clipboard
Pediatric hemophagocytic syndromes: a diagnostic and therapeutic challenge
Allergy Asthma Clin Immunol.
.
Abstract
Pediatric hemophagocytic syndrome (HS) is a severe and often fatal clinical disorder. This syndrome is frequently unrecognized, and thus, affected children may receive suboptimal management, leading to an increase in mortality. The purpose of this review is to provide a clinical guide to (1) the recognition of HS based on clinical, biologic, and pathologic features; (2) the identification of the primary cause of HS in a given affected child; and (3) the initiation of effective treatment in a timely manner.
Figures
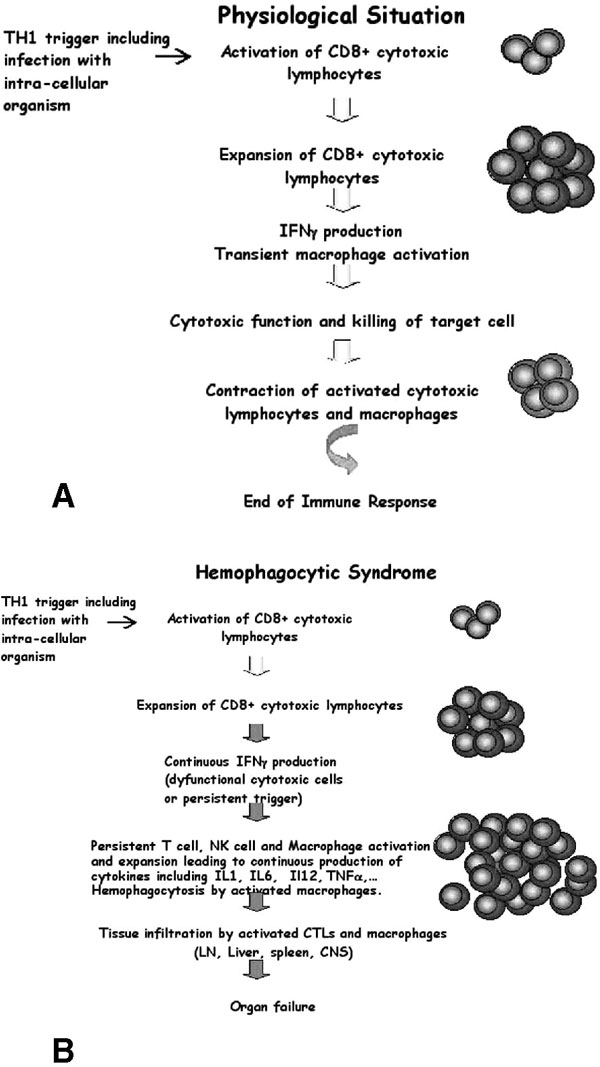
Schematic overview of antigen specific CD8+ T-cell response in a normal individual (A) and in a patient with hemophagocytic syndrome (B). In response to an infectious trigger, antigen-specific CD8+ T cells transiently undergo massive expansion, use cell-mediated cytolysis, and produce interferon-γ (IFN-γ). After pathogen clearance, this immune response is self-limiting and most cells die, leaving a reduced number of memory T and B cells. During the course of hemophagocytic syndrome, uncontrolled expansion of antigen-specific effectors occurs. Activated lymphocytes secrete high levels of INF-γ and induce a feedback loop on macrophage and T cells, which continuously activate each other and expand. High levels of inflammatory cytokines are secreted, including IFNγ, tumour necrosis factor-α, interleukin (IL)-1, IL-6, and IL-18. Activated macrophages phagocytose bystander hematopoietic cells (hemophagocytosis). Activated lymphocytes and macrophages infiltrate various organs, resulting in massive tissue necrosis and organ failure.
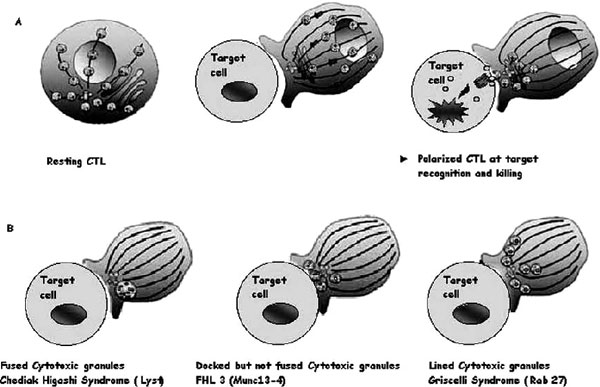
Cytotoxic granules in wild-type cytotoxic T lymphocytes (CTLs) and in CTLs from patients with genetic defects. A, Illustrations of the distribution of cytotoxic granules on microtubules (lines) in a resting human CTL (left panel). Perforin and granzyme are represented as red and green circles inside granules; one granule of each only is shown for clarity. After a CTL encounters a target cell, cytotoxic granules polarize and move along microtubules (middle panel) to the microtubule organizing centre (in blue), which migrates to the immunologic synapse and induces apoptosis of the target cell after the endocytosis of cytotoxic granules in its cytoplasm (right panel). B, Illustration of images of CTLs from patients lacking Lyst (Chédiak-Higashi syndrome), MUNC13-4 (FHL3), or RAB27A (Griscelli syndrome 2) conjugated with target cells.
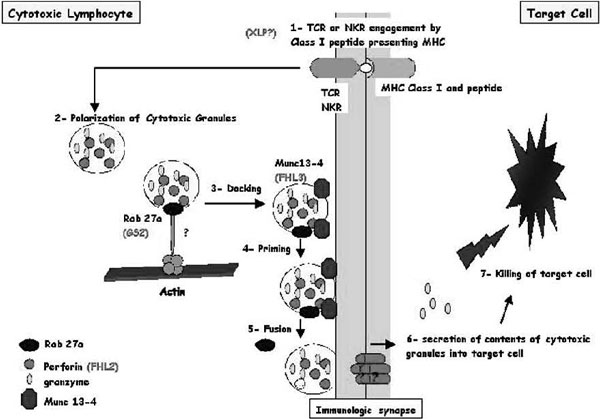
Schematic representation of cytotoxic granule exocytosis and target killing following target recognition by cytotoxic T lymphocytes (CTLs) or natural killer (NK) cells) Recognition of a peptide-major histocompatibility complex class I molecule presented by a target cell induces activation of cytotoxic lymphocytes (CTLs and NK cells). After cell conjugate formation, activated lymphocytes polarize their lytic granules toward the cell-to-cell contact, organized as an immunologic synapse. RAB27A is expected to promote the terminal transport and/or the docking step of the cytotoxic granules at the immunologic synapse. For its function, RAB27A potentially associates with unknown effectors and with MUNC13-4. MUNC13-4 functions as a priming factor, allowing cytotoxic granules to reach a fusion-competent state before membrane fusion and granule secretion occur. In 30% of patients with familial hemophagocytic lymphohistiocytosis (FHL), cytotoxic granules are defective in their functional perforin content (FHL2); in another 30% of the patients, cytotoxic granules are defective in their priming state and thus secretion (FHL3). Defective RAB27A in patients with Griscelli syndrome 2 impairs terminal transport and thus exocytosis of the lytic granule contents. X-linked lymphoproliferation and polymerization of perforin are represented with a question mark because there is no experimental proof that they act as represented in this scheme.
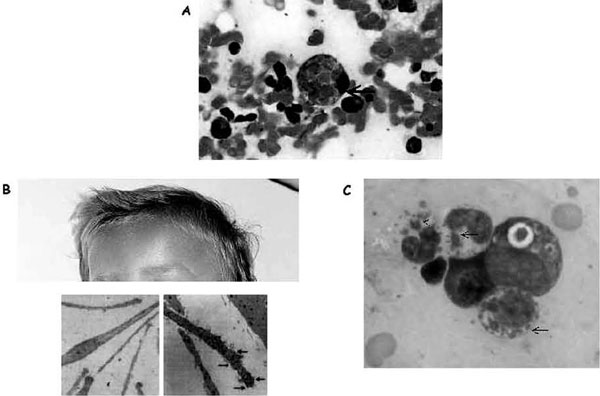
Illustration of hemophagocytosis and the most prominent extrahematologic features of Griscelli and Chédiak-Higashi syndromes. A, Hemophagocytosis in the bone marrow of a patient with familial hemophagocytic lymphohistiocytosis; arrow indicates an activated macrophage that has ingested several red blood cells. B, Partial view of the head of a child with Griscelli syndrome 2, shown to emphasize the ashengrey colour of hair. Electron microscopy images of a normal hair (left panel) and a hair of a person with Griscelli syndrome (right panel) are shown below; arrows indicate clumps of melanin specific for this disease. A defect in any of the proteins (myosin Va, RAB27A, or melanophilin) leads to identical pigmentary dilution in the three forms of Griscelli syndrome and their mouse models. C, Blood smear taken from a patient with Chédiak-Higashi syndrome. Arrows indicate large granules present in all cell lineages that orient the diagnosis toward Chédiak-Higashi syndrome.
Similar articles
-
Adult cancer-related hemophagocytic lymphohistiocytosis - a challenging diagnosis: a case report.J Med Case Rep. 2017 Jun 27;11(1):172. doi: 10.1186/s13256-017-1344-x. J Med Case Rep. 2017. PMID: 28651636 Free PMC article.
-
When T cells and macrophages do not talk: the hemophagocytic syndromes.Curr Opin Hematol. 2008 Jul;15(4):359-67. doi: 10.1097/MOH.0b013e3282f97f88. Curr Opin Hematol. 2008. PMID: 18536575 Review.
-
[Hemophagocytic syndrome: diagnostic problems].Przegl Lek. 2006;63(1):47-52. Przegl Lek. 2006. PMID: 16892901 Review. Polish.
-
High-Volume Hemofiltration in Critically Ill Patients With Secondary Hemophagocytic Lymphohistiocytosis/Macrophage Activation Syndrome: A Prospective Study in the PICU.Pediatr Crit Care Med. 2016 Oct;17(10):e437-e443. doi: 10.1097/PCC.0000000000000896. Pediatr Crit Care Med. 2016. PMID: 27487914 Clinical Trial.
-
Consensus recommendations for the diagnosis and management of hemophagocytic lymphohistiocytosis associated with malignancies.Haematologica. 2015 Aug;100(8):997-1004. doi: 10.3324/haematol.2015.123562. Haematologica. 2015. PMID: 26314082 Free PMC article.
Cited by
-
Hemophagocytic syndrome with atypical presentation in an adolescent.BMJ Case Rep. 2013 Sep 11;2013:bcr2013200929. doi: 10.1136/bcr-2013-200929. BMJ Case Rep. 2013. PMID: 24027258 Free PMC article.
-
Clinico-laboratory profile and perforin gene mutations of pediatric hemophagocytic lymphohistiocytosis cases: a five-year single center study.Pan Afr Med J. 2020 Aug 27;36:354. doi: 10.11604/pamj.2020.36.354.25079. eCollection 2020. Pan Afr Med J. 2020. PMID: 33224420 Free PMC article.
-
Debate around infection-dependent hemophagocytic syndrome in paediatrics.BMC Infect Dis. 2013 Jan 16;13:15. doi: 10.1186/1471-2334-13-15. BMC Infect Dis. 2013. PMID: 23324497 Free PMC article. Review.
-
[Macrophage activation syndrome complicating family lymphohistiocytosis].Pan Afr Med J. 2017 Feb 24;26:93. doi: 10.11604/pamj.2017.26.93.6235. eCollection 2017. Pan Afr Med J. 2017. PMID: 28491224 Free PMC article. French.
References
-
- Scott R, Robb-Smith A. Histiocytic medullary reticulosis. Lancet. 1939;2:194–8.
-
- Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18:29–33. - PubMed
-
- Stephan JL, Zeller J, Hubert P. Macrophage activation syndrome and rheumatic disease in childhood: a report of four new cases. Clin Exp Rheumatol. 1993;11:451–6. - PubMed
LinkOut - more resources
Full Text Sources
