

Robust unmixing of tumor states in array comparative genomic hybridization data
- PMID: 20529894
- PMCID: PMC2881397
- DOI: 10.1093/bioinformatics/btq213
Robust unmixing of tumor states in array comparative genomic hybridization data
Abstract
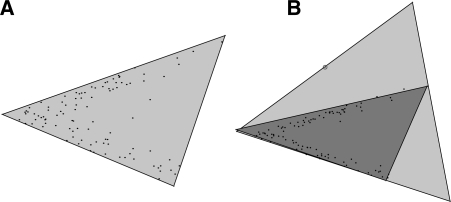
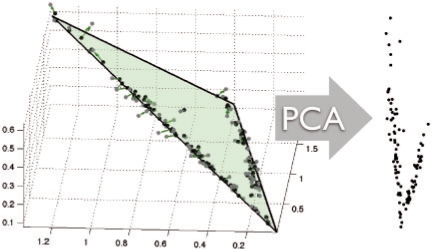
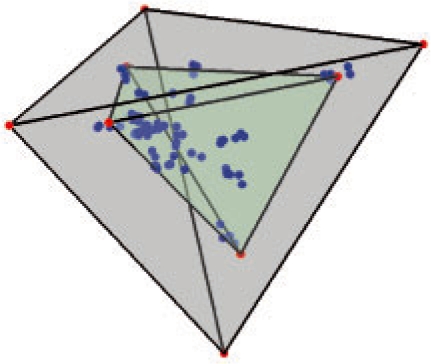
Motivation: Tumorigenesis is an evolutionary process by which tumor cells acquire sequences of mutations leading to increased growth, invasiveness and eventually metastasis. It is hoped that by identifying the common patterns of mutations underlying major cancer sub-types, we can better understand the molecular basis of tumor development and identify new diagnostics and therapeutic targets. This goal has motivated several attempts to apply evolutionary tree reconstruction methods to assays of tumor state. Inference of tumor evolution is in principle aided by the fact that tumors are heterogeneous, retaining remnant populations of different stages along their development along with contaminating healthy cell populations. In practice, though, this heterogeneity complicates interpretation of tumor data because distinct cell types are conflated by common methods for assaying the tumor state. We previously proposed a method to computationally infer cell populations from measures of tumor-wide gene expression through a geometric interpretation of mixture type separation, but this approach deals poorly with noisy and outlier data.
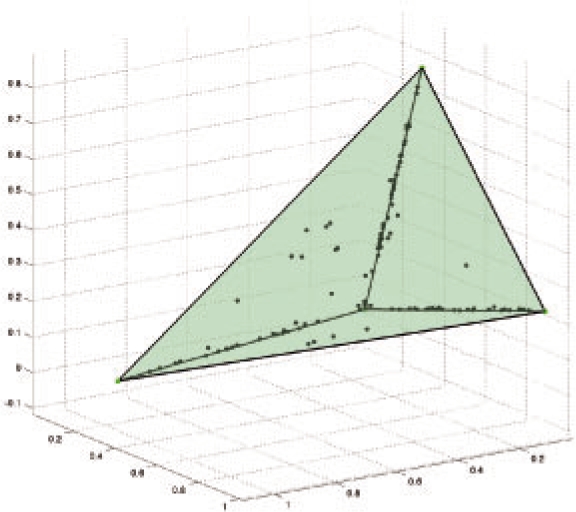
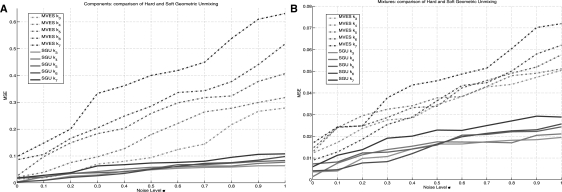
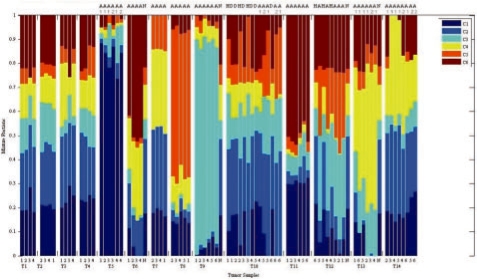
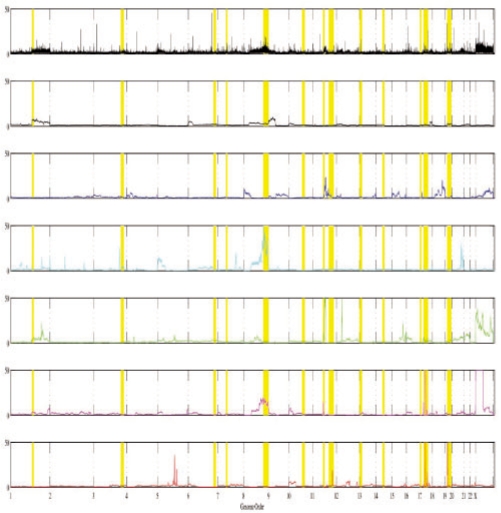
Results: In the present work, we propose a new method to perform tumor mixture separation efficiently and robustly to an experimental error. The method builds on the prior geometric approach but uses a novel objective function allowing for robust fits that greatly reduces the sensitivity to noise and outliers. We further develop an efficient gradient optimization method to optimize this 'soft geometric unmixing' objective for measurements of tumor DNA copy numbers assessed by array comparative genomic hybridization (aCGH) data. We show, on a combination of semi-synthetic and real data, that the method yields fast and accurate separation of tumor states.
Conclusions: We have shown a novel objective function and optimization method for the robust separation of tumor sub-types from aCGH data and have shown that the method provides fast, accurate reconstruction of tumor states from mixed samples. Better solutions to this problem can be expected to improve our ability to accurately identify genetic abnormalities in primary tumor samples and to infer patterns of tumor evolution.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures

References
-
- Atkins JH, Gershell LJ. From the analyst's couch: selective anticancer drugs. Nat. Rev. Cancer. 2002;2:645–646. - PubMed
-
- Beerenwinkel N, et al. Mtreemix: a software package for learning and using mixture models of mutagenetic trees. Bioinformatics. 2005;21:2106–2107. - PubMed
-
- Bild AH, et al. Opinion: linking oncogenic pathways with therapeutic opportunities. Nat. Rev. Cancer. 2006;6:735–741. - PubMed
-
- Boyd S, Vandenberghe L. Convex Optimization. New York, NY: Cambridge University Press; 2004.
-
- Chan T, et al. A convex analysis based minimum-volume enclosing simplex algorithm for hyperspectral unmixing. IEEE Trans. Signal Proc. 2009;57:4418–4432.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
