

Optimal algorithms for haplotype assembly from whole-genome sequence data
- PMID: 20529904
- PMCID: PMC2881399
- DOI: 10.1093/bioinformatics/btq215
Optimal algorithms for haplotype assembly from whole-genome sequence data
Abstract
Motivation: Haplotype inference is an important step for many types of analyses of genetic variation in the human genome. Traditional approaches for obtaining haplotypes involve collecting genotype information from a population of individuals and then applying a haplotype inference algorithm. The development of high-throughput sequencing technologies allows for an alternative strategy to obtain haplotypes by combining sequence fragments. The problem of 'haplotype assembly' is the problem of assembling the two haplotypes for a chromosome given the collection of such fragments, or reads, and their locations in the haplotypes, which are pre-determined by mapping the reads to a reference genome. Errors in reads significantly increase the difficulty of the problem and it has been shown that the problem is NP-hard even for reads of length 2. Existing greedy and stochastic algorithms are not guaranteed to find the optimal solutions for the haplotype assembly problem.
Results: In this article, we proposed a dynamic programming algorithm that is able to assemble the haplotypes optimally with time complexity O(m x 2(k) x n), where m is the number of reads, k is the length of the longest read and n is the total number of SNPs in the haplotypes. We also reduce the haplotype assembly problem into the maximum satisfiability problem that can often be solved optimally even when k is large. Taking advantage of the efficiency of our algorithm, we perform simulation experiments demonstrating that the assembly of haplotypes using reads of length typical of the current sequencing technologies is not practical. However, we demonstrate that the combination of this approach and the traditional haplotype phasing approaches allow us to practically construct haplotypes containing both common and rare variants.
Figures
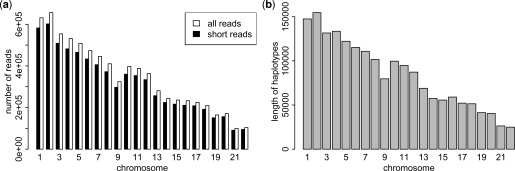
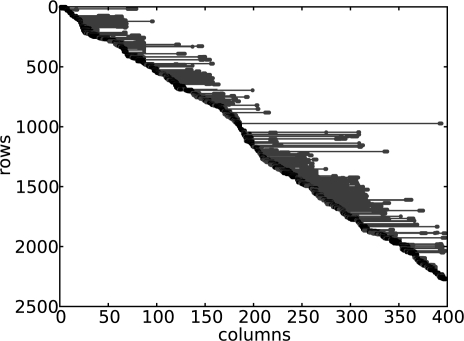
Similar articles
-
Hap-seq: an optimal algorithm for haplotype phasing with imputation using sequencing data.J Comput Biol. 2013 Feb;20(2):80-92. doi: 10.1089/cmb.2012.0091. J Comput Biol. 2013. PMID: 23383995 Free PMC article.
-
Exact algorithms for haplotype assembly from whole-genome sequence data.Bioinformatics. 2013 Aug 15;29(16):1938-45. doi: 10.1093/bioinformatics/btt349. Epub 2013 Jun 18. Bioinformatics. 2013. PMID: 23782612
-
Efficient algorithms for polyploid haplotype phasing.BMC Genomics. 2018 May 9;19(Suppl 2):110. doi: 10.1186/s12864-018-4464-9. BMC Genomics. 2018. PMID: 29764364 Free PMC article.
-
Haplotype inference by Pure Parsimony: a survey.J Comput Biol. 2010 Aug;17(8):969-92. doi: 10.1089/cmb.2009.0101. J Comput Biol. 2010. PMID: 20726791 Review.
-
Towards accurate, contiguous and complete alignment-based polyploid phasing algorithms.Genomics. 2022 May;114(3):110369. doi: 10.1016/j.ygeno.2022.110369. Epub 2022 Apr 26. Genomics. 2022. PMID: 35483655 Review.
Cited by
-
Haplotype-resolved whole-genome sequencing by contiguity-preserving transposition and combinatorial indexing.Nat Genet. 2014 Dec;46(12):1343-9. doi: 10.1038/ng.3119. Epub 2014 Oct 19. Nat Genet. 2014. PMID: 25326703 Free PMC article.
-
An accurate algorithm for the detection of DNA fragments from dilution pool sequencing experiments.Bioinformatics. 2018 Jan 1;34(1):155-162. doi: 10.1093/bioinformatics/btx436. Bioinformatics. 2018. PMID: 29036419 Free PMC article.
-
PhISCS: a combinatorial approach for subperfect tumor phylogeny reconstruction via integrative use of single-cell and bulk sequencing data.Genome Res. 2019 Nov;29(11):1860-1877. doi: 10.1101/gr.234435.118. Epub 2019 Oct 18. Genome Res. 2019. PMID: 31628256 Free PMC article.
-
Allele Phasing Greatly Improves the Phylogenetic Utility of Ultraconserved Elements.Syst Biol. 2019 Jan 1;68(1):32-46. doi: 10.1093/sysbio/syy039. Syst Biol. 2019. PMID: 29771371 Free PMC article.
-
The next phase in human genetics.Nat Biotechnol. 2011 Jan;29(1):38-9. doi: 10.1038/nbt.1757. Nat Biotechnol. 2011. PMID: 21221098 No abstract available.
References
-
- 1000 Genomes Project. A deep catalog of human genetic variation. 2010 Available at http://www.1000genomes.org/ (last accessed date April 23, 2010)
-
- Bansal V, Bafna V. HapCUT: an efficient and accurate algorithm for the haplotype assembly problem. Bioinformatics. 2008;24:i153. - PubMed
-
- Biere A, et al. Frontiers in Artificial Intelligence and Applications. Vol. 185. Nieume Hemweg, Amsterdam: IOS Press; 2009. Handbook of Satisfiability.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
