

Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM
- PMID: 20529912
- PMCID: PMC2881367
- DOI: 10.1093/bioinformatics/btq182
Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM
Abstract
Motivation: High-throughput data is providing a comprehensive view of the molecular changes in cancer tissues. New technologies allow for the simultaneous genome-wide assay of the state of genome copy number variation, gene expression, DNA methylation and epigenetics of tumor samples and cancer cell lines. Analyses of current data sets find that genetic alterations between patients can differ but often involve common pathways. It is therefore critical to identify relevant pathways involved in cancer progression and detect how they are altered in different patients.
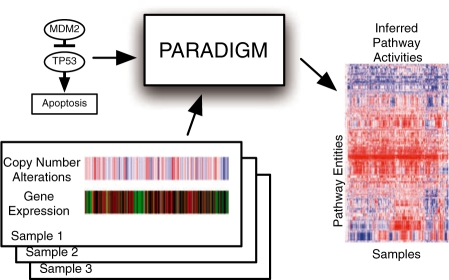
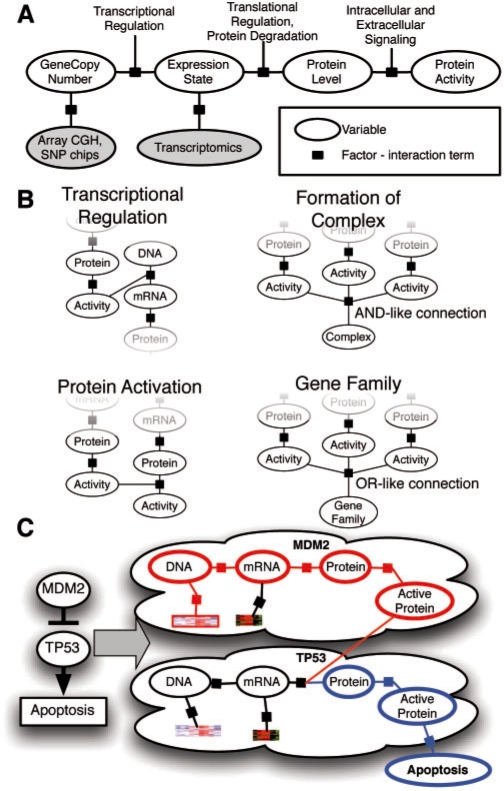
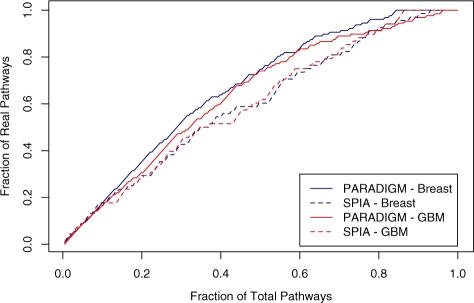
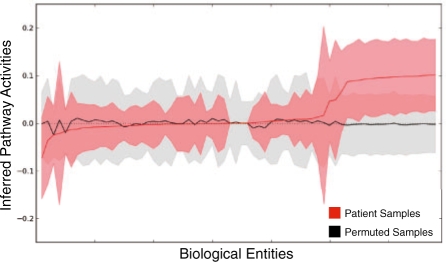
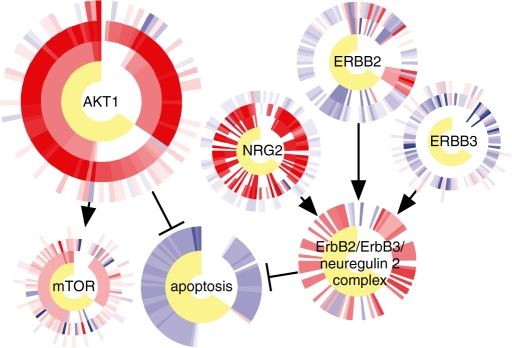
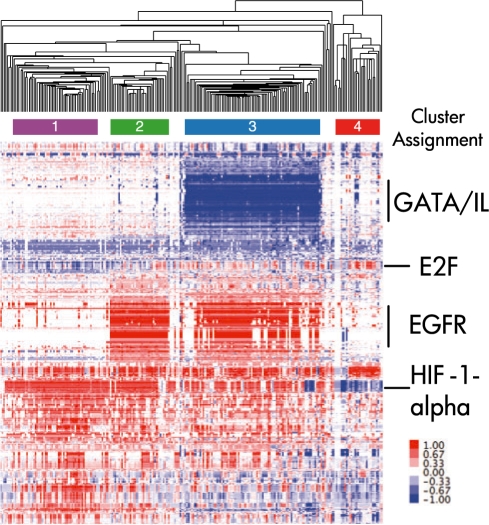
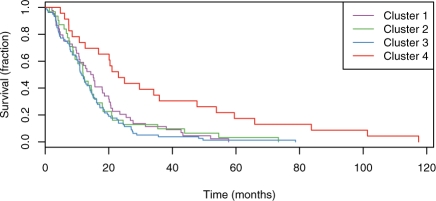
Results: We present a novel method for inferring patient-specific genetic activities incorporating curated pathway interactions among genes. A gene is modeled by a factor graph as a set of interconnected variables encoding the expression and known activity of a gene and its products, allowing the incorporation of many types of omic data as evidence. The method predicts the degree to which a pathway's activities (e.g. internal gene states, interactions or high-level 'outputs') are altered in the patient using probabilistic inference. Compared with a competing pathway activity inference approach called SPIA, our method identifies altered activities in cancer-related pathways with fewer false-positives in both a glioblastoma multiform (GBM) and a breast cancer dataset. PARADIGM identified consistent pathway-level activities for subsets of the GBM patients that are overlooked when genes are considered in isolation. Further, grouping GBM patients based on their significant pathway perturbations divides them into clinically-relevant subgroups having significantly different survival outcomes. These findings suggest that therapeutics might be chosen that target genes at critical points in the commonly perturbed pathway(s) of a group of patients.
Availability: Source code available at http://sbenz.github.com/Paradigm,.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
References
-
- Alizadeh AA, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. - PubMed
-
- Allison DB, et al. Microarray data analysis: from disarray to consolidation and consensus. Nat. Rev. Genet. 2006;7:55–65. - PubMed
-
- Beer MA, Tavazoie S. Predicting gene expression from sequence. Cell. 2004;117:185–198. - PubMed
-
- BioPAX working group. BioPAX–biological pathways exchange language. Documentation. 2004
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
