

Markov dynamic models for long-timescale protein motion
- PMID: 20529916
- PMCID: PMC2881362
- DOI: 10.1093/bioinformatics/btq177
Markov dynamic models for long-timescale protein motion
Abstract
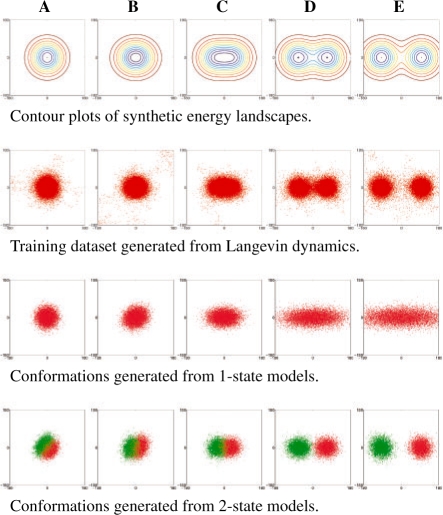
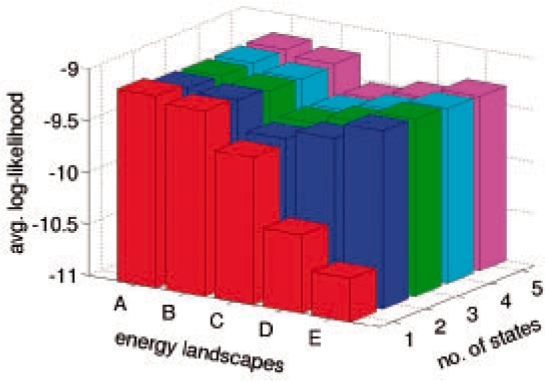
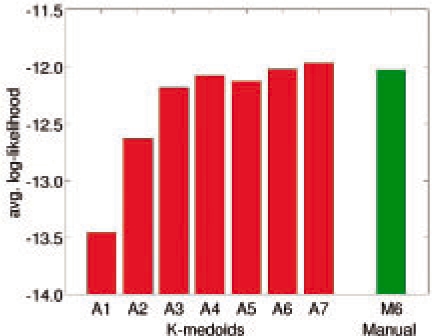
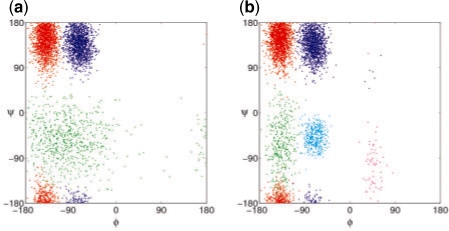
Molecular dynamics (MD) simulation is a well-established method for studying protein motion at the atomic scale. However, it is computationally intensive and generates massive amounts of data. One way of addressing the dual challenges of computation efficiency and data analysis is to construct simplified models of long-timescale protein motion from MD simulation data. In this direction, we propose to use Markov models with hidden states, in which the Markovian states represent potentially overlapping probabilistic distributions over protein conformations. We also propose a principled criterion for evaluating the quality of a model by its ability to predict long-timescale protein motions. Our method was tested on 2D synthetic energy landscapes and two extensively studied peptides, alanine dipeptide and the villin headpiece subdomain (HP-35 NleNle). One interesting finding is that although a widely accepted model of alanine dipeptide contains six states, a simpler model with only three states is equally good for predicting long-timescale motions. We also used the constructed Markov models to estimate important kinetic and dynamic quantities for protein folding, in particular, mean first-passage time. The results are consistent with available experimental measurements.
Figures
Similar articles
-
Progress and challenges in the automated construction of Markov state models for full protein systems.J Chem Phys. 2009 Sep 28;131(12):124101. doi: 10.1063/1.3216567. J Chem Phys. 2009. PMID: 19791846 Free PMC article.
-
Tutorial on how to build non-Markovian dynamic models from molecular dynamics simulations for studying protein conformational changes.J Chem Phys. 2024 Mar 28;160(12):121501. doi: 10.1063/5.0189429. J Chem Phys. 2024. PMID: 38516972 Free PMC article.
-
On the advantages of exploiting memory in Markov state models for biomolecular dynamics.J Chem Phys. 2020 Jul 7;153(1):014105. doi: 10.1063/5.0010787. J Chem Phys. 2020. PMID: 32640825
-
Markov state models provide insights into dynamic modulation of protein function.Acc Chem Res. 2015 Feb 17;48(2):414-22. doi: 10.1021/ar5002999. Epub 2015 Jan 3. Acc Chem Res. 2015. PMID: 25625937 Free PMC article. Review.
-
Simulating the peptide folding kinetic related spectra based on the Markov State Model.Adv Exp Med Biol. 2014;805:199-220. doi: 10.1007/978-3-319-02970-2_9. Adv Exp Med Biol. 2014. PMID: 24446363 Review.
Cited by
-
Computational models of protein kinematics and dynamics: beyond simulation.Annu Rev Anal Chem (Palo Alto Calif). 2012;5:273-91. doi: 10.1146/annurev-anchem-062011-143024. Epub 2012 Apr 9. Annu Rev Anal Chem (Palo Alto Calif). 2012. PMID: 22524225 Free PMC article. Review.
-
QAARM: quasi-anharmonic autoregressive model reveals molecular recognition pathways in ubiquitin.Bioinformatics. 2011 Jul 1;27(13):i52-60. doi: 10.1093/bioinformatics/btr248. Bioinformatics. 2011. PMID: 21685101 Free PMC article.
-
Quantifying the Sources of Kinetic Frustration in Folding Simulations of Small Proteins.J Chem Theory Comput. 2014 Aug 12;10(8):2964-2974. doi: 10.1021/ct500361w. Epub 2014 Jun 13. J Chem Theory Comput. 2014. PMID: 25136267 Free PMC article.
-
SIMS: a hybrid method for rapid conformational analysis.PLoS One. 2013 Jul 23;8(7):e68826. doi: 10.1371/journal.pone.0068826. Print 2013. PLoS One. 2013. PMID: 23935893 Free PMC article.
References
-
- Amadei A, et al. Essential dynamics of proteins. Prot. Struct. Funct. Genet. 1993;17:412–425. - PubMed
-
- Amato NM, et al. Using motion planning to map protein folding landscapes and analyze folding kinetics of known native structures. J. Comput. Biol. 2003;10:239–255. - PubMed
-
- Apaydin MS, et al. Stochastic roadmap simulation: an efficient representation and algorithm for analyzing molecular motion. J. Comput. Biol. 2003;10:257–281. - PubMed
-
- Bishop G. Pattern Recognition and Machine Learning. New York: Springer; 2007.
-
- Chekmarev DS, et al. Long-time conformational transitions of alanine dipeptide in aqueous solution: continuous and discrete-state kinetic models. J. Phys. Chem. B. 2004;108:19487–19495.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
