

Fragment-free approach to protein folding using conditional neural fields
- PMID: 20529922
- PMCID: PMC2881378
- DOI: 10.1093/bioinformatics/btq193
Fragment-free approach to protein folding using conditional neural fields
Abstract
Motivation: One of the major bottlenecks with ab initio protein folding is an effective conformation sampling algorithm that can generate native-like conformations quickly. The popular fragment assembly method generates conformations by restricting the local conformations of a protein to short structural fragments in the PDB. This method may limit conformations to a subspace to which the native fold does not belong because (i) a protein with really new fold may contain some structural fragments not in the PDB and (ii) the discrete nature of fragments may prevent them from building a native-like fold. Previously we have developed a conditional random fields (CRF) method for fragment-free protein folding that can sample conformations in a continuous space and demonstrated that this CRF method compares favorably to the popular fragment assembly method. However, the CRF method is still limited by its capability of generating conformations compatible with a sequence.
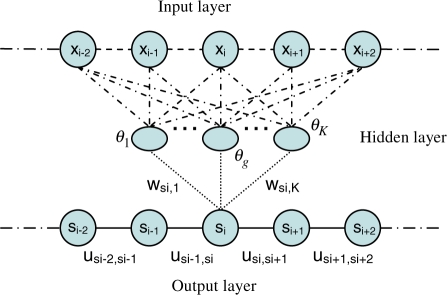
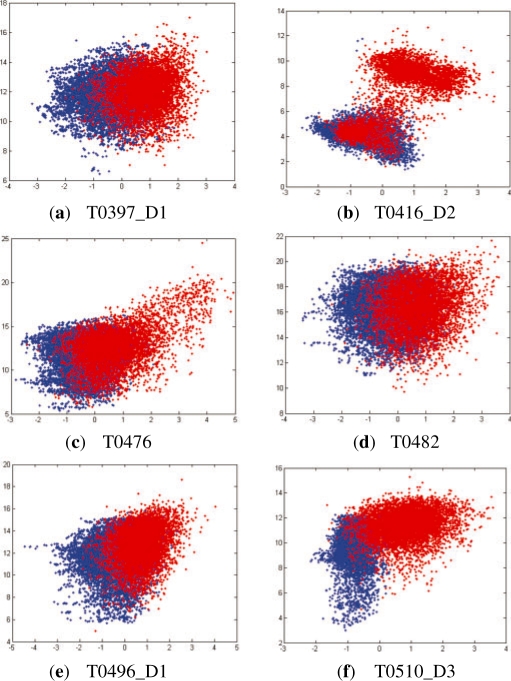
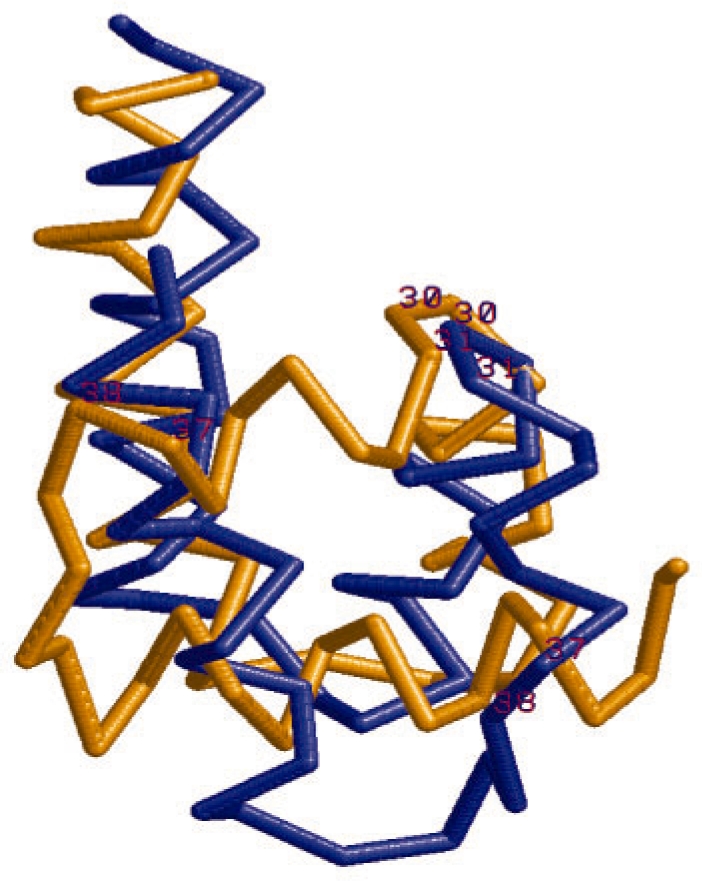
Results: We present a new fragment-free approach to protein folding using a recently invented probabilistic graphical model conditional neural fields (CNF). This new CNF method is much more powerful than CRF in modeling the sophisticated protein sequence-structure relationship and thus, enables us to generate native-like conformations more easily. We show that when coupled with a simple energy function and replica exchange Monte Carlo simulation, our CNF method can generate decoys much better than CRF on a variety of test proteins including the CASP8 free-modeling targets. In particular, our CNF method can predict a correct fold for T0496_D1, one of the two CASP8 targets with truly new fold. Our predicted model for T0496 is significantly better than all the CASP8 models.
Figures

Similar articles
-
A probabilistic and continuous model of protein conformational space for template-free modeling.J Comput Biol. 2010 Jun;17(6):783-98. doi: 10.1089/cmb.2009.0235. J Comput Biol. 2010. PMID: 20583926 Free PMC article.
-
A Probabilistic Graphical Model for Ab Initio Folding.Res Comput Mol Biol. 2009;5541:59-73. doi: 10.1007/978-3-642-02008-7_5. Res Comput Mol Biol. 2009. PMID: 23459639 Free PMC article.
-
A conditional random fields method for RNA sequence-structure relationship modeling and conformation sampling.Bioinformatics. 2011 Jul 1;27(13):i102-10. doi: 10.1093/bioinformatics/btr232. Bioinformatics. 2011. PMID: 21685058 Free PMC article.
-
3DRobot: automated generation of diverse and well-packed protein structure decoys.Bioinformatics. 2016 Feb 1;32(3):378-87. doi: 10.1093/bioinformatics/btv601. Epub 2015 Oct 14. Bioinformatics. 2016. PMID: 26471454 Free PMC article.
-
A homology/ab initio hybrid algorithm for sampling near-native protein conformations.J Comput Chem. 2013 Aug 15;34(22):1925-36. doi: 10.1002/jcc.23339. Epub 2013 Jun 3. J Comput Chem. 2013. PMID: 23728619
Cited by
-
Alignment of distantly related protein structures: algorithm, bound and implications to homology modeling.Bioinformatics. 2011 Sep 15;27(18):2537-45. doi: 10.1093/bioinformatics/btr432. Epub 2011 Jul 26. Bioinformatics. 2011. PMID: 21791532 Free PMC article.
-
Representations of protein structure for exploring the conformational space: A speed-accuracy trade-off.Comput Struct Biotechnol J. 2021 Apr 28;19:2618-2625. doi: 10.1016/j.csbj.2021.04.049. eCollection 2021. Comput Struct Biotechnol J. 2021. PMID: 34025948 Free PMC article.
-
A conditional neural fields model for protein threading.Bioinformatics. 2012 Jun 15;28(12):i59-66. doi: 10.1093/bioinformatics/bts213. Bioinformatics. 2012. PMID: 22689779 Free PMC article.
-
Predicting the molecular interactions of CRIP1a-cannabinoid 1 receptor with integrated molecular modeling approaches.Bioorg Med Chem Lett. 2014 Feb 15;24(4):1158-65. doi: 10.1016/j.bmcl.2013.12.119. Epub 2014 Jan 8. Bioorg Med Chem Lett. 2014. PMID: 24461351 Free PMC article.
-
AcconPred: Predicting Solvent Accessibility and Contact Number Simultaneously by a Multitask Learning Framework under the Conditional Neural Fields Model.Biomed Res Int. 2015;2015:678764. doi: 10.1155/2015/678764. Epub 2015 Aug 3. Biomed Res Int. 2015. PMID: 26339631 Free PMC article.
References
-
- Aarts E, Korst J. Simulated Annealing and Boltzmann Machines: A Stochastic Approach to Combinatorial Optimization and Neural Computing. New York: Wiley; 1991.
-
- Branden C.-I, Tooze J. Introduction to Protein Structure. New York, London: Garland Publishing; 1999.
-
- Chen WW, et al. A knowledge-based move set for protein folding. Proteins-Struct. Funct. Bioinformatics. 2007;66:682–688. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
