

A statistical method for the detection of variants from next-generation resequencing of DNA pools
- PMID: 20529923
- PMCID: PMC2881398
- DOI: 10.1093/bioinformatics/btq214
A statistical method for the detection of variants from next-generation resequencing of DNA pools
Erratum in
-
A statistical method for the detection of variants from next-generation resequencing of DNA pools.Bioinformatics. 2016 Oct 15;32(20):3213. doi: 10.1093/bioinformatics/btw520. Epub 2016 Aug 29. Bioinformatics. 2016. PMID: 27578802 Free PMC article. No abstract available.
Abstract
Motivation: Next-generation sequencing technologies have enabled the sequencing of several human genomes in their entirety. However, the routine resequencing of complete genomes remains infeasible. The massive capacity of next-generation sequencers can be harnessed for sequencing specific genomic regions in hundreds to thousands of individuals. Sequencing-based association studies are currently limited by the low level of multiplexing offered by sequencing platforms. Pooled sequencing represents a cost-effective approach for studying rare variants in large populations. To utilize the power of DNA pooling, it is important to accurately identify sequence variants from pooled sequencing data. Detection of rare variants from pooled sequencing represents a different challenge than detection of variants from individual sequencing.
Results: We describe a novel statistical approach, CRISP [Comprehensive Read analysis for Identification of Single Nucleotide Polymorphisms (SNPs) from Pooled sequencing] that is able to identify both rare and common variants by using two approaches: (i) comparing the distribution of allele counts across multiple pools using contingency tables and (ii) evaluating the probability of observing multiple non-reference base calls due to sequencing errors alone. Information about the distribution of reads between the forward and reverse strands and the size of the pools is also incorporated within this framework to filter out false variants. Validation of CRISP on two separate pooled sequencing datasets generated using the Illumina Genome Analyzer demonstrates that it can detect 80-85% of SNPs identified using individual sequencing while achieving a low false discovery rate (3-5%). Comparison with previous methods for pooled SNP detection demonstrates the significantly lower false positive and false negative rates for CRISP.
Availability: Implementation of this method is available at http://polymorphism.scripps.edu/~vbansal/software/CRISP/.
Figures


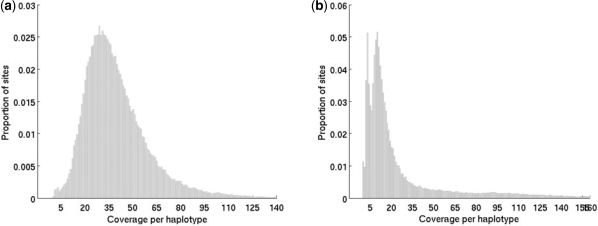
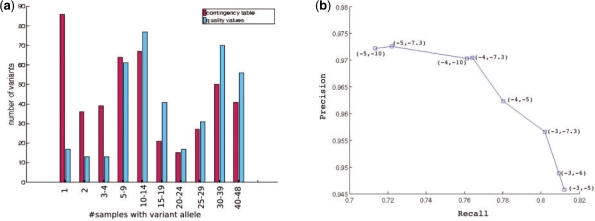
References
-
- Chernoff H. A measure of asymptotic efficiency for tests of a hypothesis based on the sum of observations. Ann. Math. Stat. 1952;23:493–507.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
