

Assessing the functional coherence of gene sets with metrics based on the Gene Ontology graph
- PMID: 20529941
- PMCID: PMC2881388
- DOI: 10.1093/bioinformatics/btq203
Assessing the functional coherence of gene sets with metrics based on the Gene Ontology graph
Abstract
Motivation: The results of initial analyses for many high-throughput technologies commonly take the form of gene or protein sets, and one of the ensuing tasks is to evaluate the functional coherence of these sets. The study of gene set function most commonly makes use of controlled vocabulary in the form of ontology annotations. For a given gene set, the statistical significance of observing these annotations or 'enrichment' may be tested using a number of methods. Instead of testing for significance of individual terms, this study is concerned with the task of assessing the global functional coherence of gene sets, for which novel metrics and statistical methods have been devised.
Results: The metrics of this study are based on the topological properties of graphs comprised of genes and their Gene Ontology annotations. A novel aspect of these methods is that both the enrichment of annotations and the relationships among annotations are considered when determining the significance of functional coherence. We applied our methods to perform analyses on an existing database and on microarray experimental results. Here, we demonstrated that our approach is highly discriminative in terms of differentiating coherent gene sets from random ones and that it provides biologically sensible evaluations in microarray analysis. We further used examples to show the utility of graph visualization as a tool for studying the functional coherence of gene sets.
Availability: The implementation is provided as a freely accessible web application at: http://projects.dbbe.musc.edu/gosteiner. Additionally, the source code written in the Python programming language, is available under the General Public License of the Free Software Foundation.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
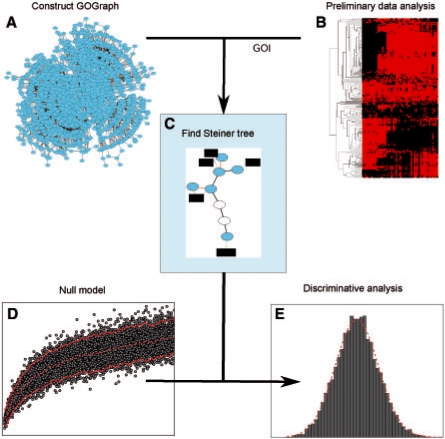
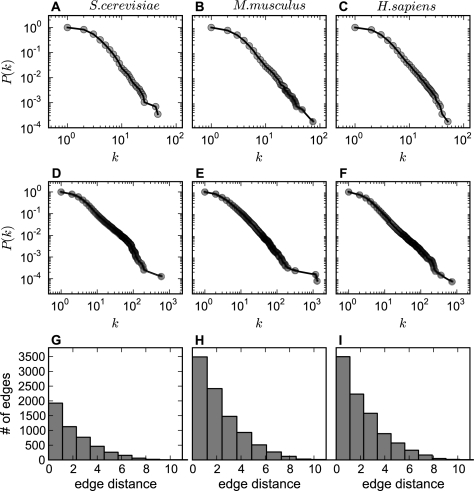
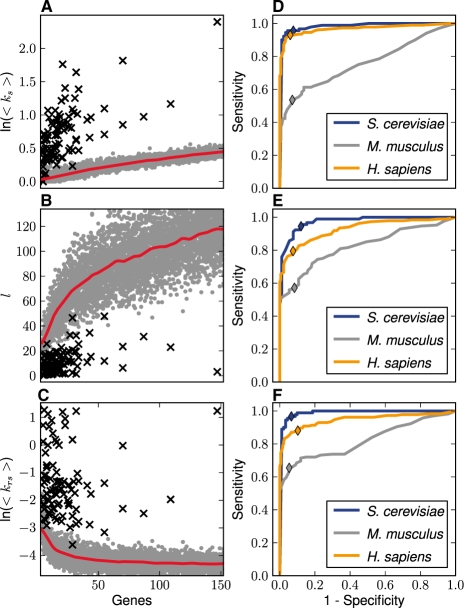
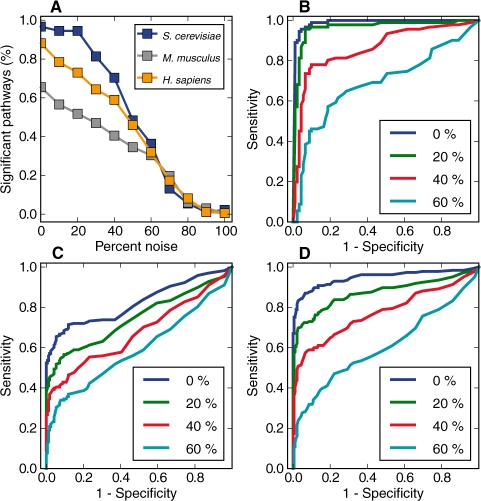
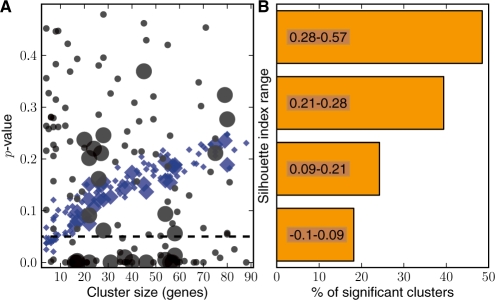
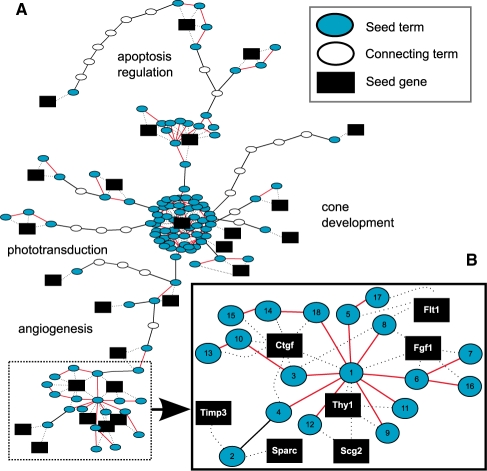
References
-
- Alexa A, et al. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. - PubMed
-
- Barabási A, Oltvai Z. Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 2004;5:101–114. - PubMed
-
- Cho R, et al. Transcriptional regulation and function during the human cell cycle. Nat. Genet. 2001;27:48–54. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
