

Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair
- PMID: 20530711
- PMCID: PMC2884153
- DOI: 10.1158/1535-7163.MCT-09-1027
Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair
Abstract
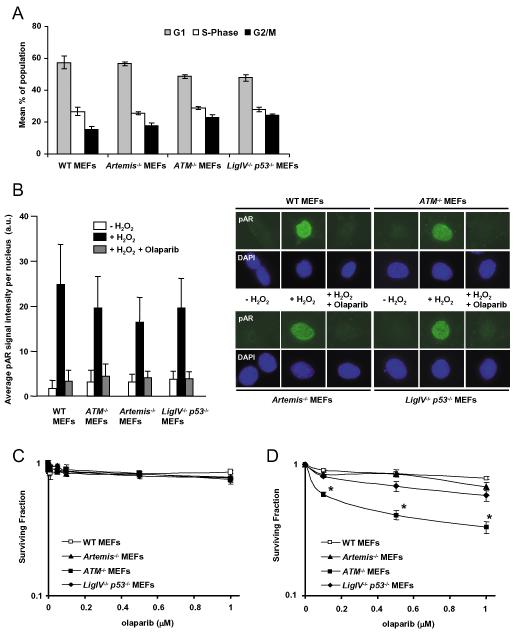
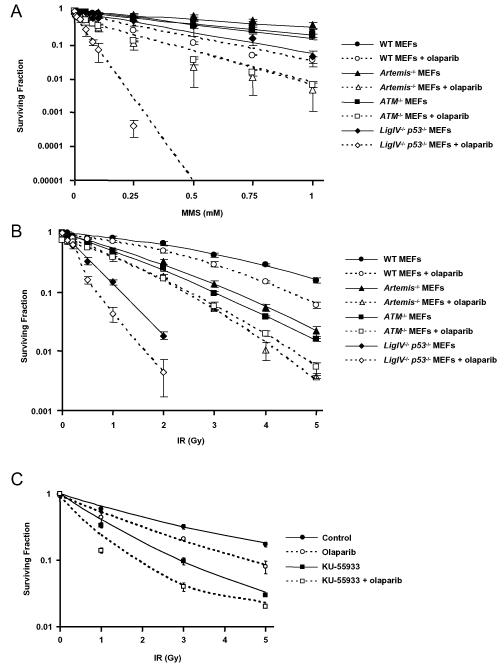
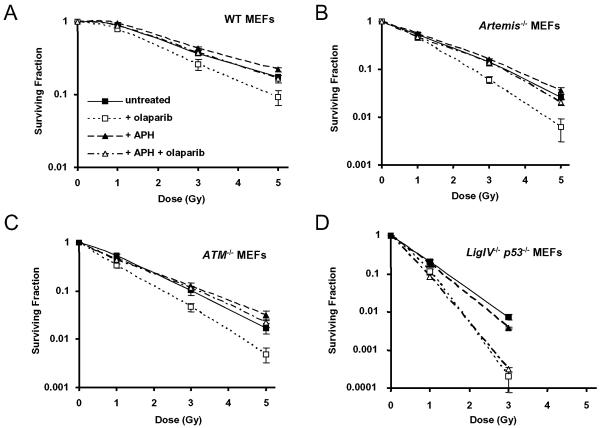
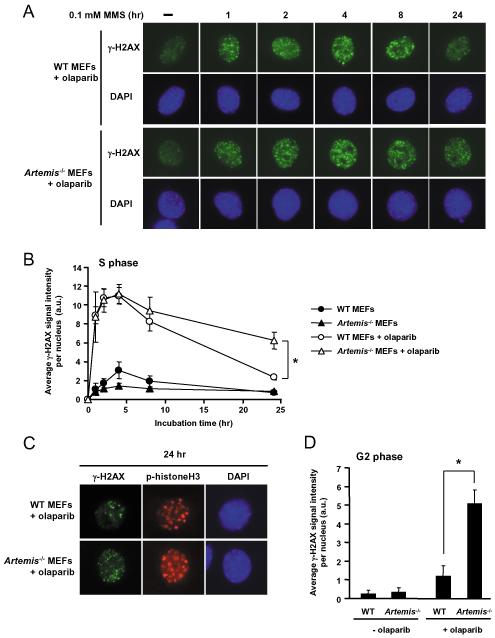
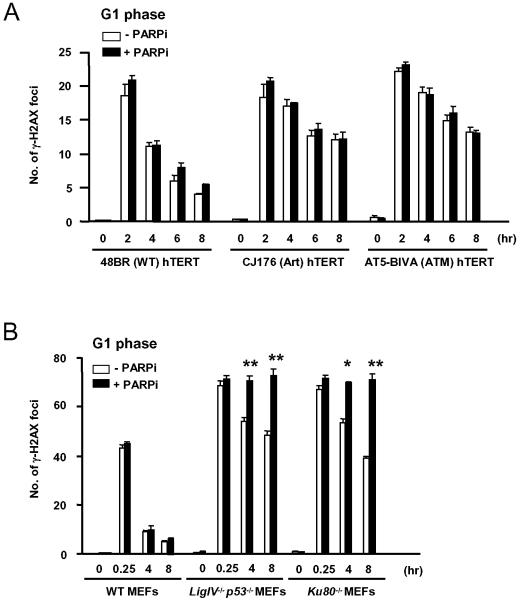
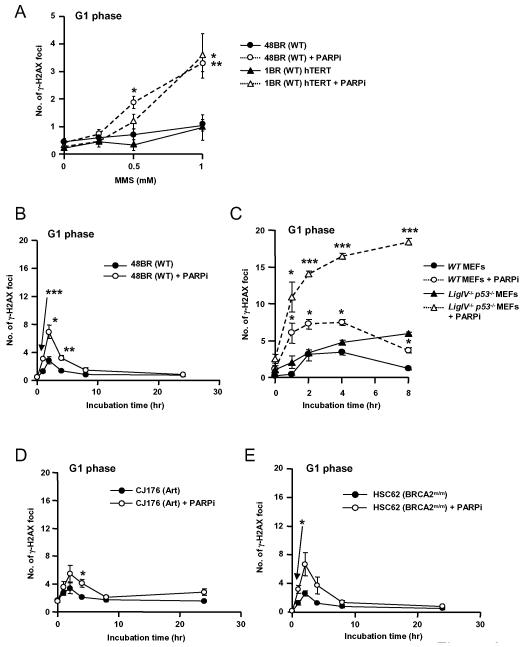
As single agents, chemical inhibitors of poly(ADP-ribose) polymerase (PARP) are nontoxic and have clinical efficacy against BRCA1- and BRCA2-deficient tumors. PARP inhibitors also enhance the cytotoxicity of ionizing radiation and alkylating agents but will only improve clinical outcomes if tumor sensitization exceeds effects on normal tissues. It is unclear how tumor DNA repair proficiency affects the degree of sensitization. We have previously shown that the radiosensitizing effect of PARP inhibition requires DNA replication and will therefore affect rapidly proliferating tumors more than normal tissues. Because many tumors exhibit defective DNA repair, we investigated the impact of double-strand break (DSB) repair integrity on the sensitizing effects of the PARP inhibitor olaparib. Sensitization to ionizing radiation and the alkylating agent methylmethane sulfonate was enhanced in DSB repair-deficient cells. In Artemis(-/-) and ATM(-/-) mouse embryo fibroblasts, sensitization was replication dependent and associated with defective repair of replication-associated damage. Radiosensitization of Ligase IV(-/-) mouse embryo fibroblasts was independent of DNA replication and is explained by inhibition of "alternative" end joining. After methylmethane sulfonate treatment, PARP inhibition promoted replication-independent accumulation of DSB, repair of which required Ligase IV. Our findings predict that the sensitizing effects of PARP inhibitors will be more pronounced in rapidly dividing and/or DNA repair defective tumors than normal tissues and show their potential to enhance the therapeutic ratio achieved by conventional DNA-damaging agents.
Figures
Similar articles
-
Alkylation DNA damage in combination with PARP inhibition results in formation of S-phase-dependent double-strand breaks.DNA Repair (Amst). 2010 Aug 5;9(8):929-36. doi: 10.1016/j.dnarep.2010.05.007. Epub 2010 Jun 22. DNA Repair (Amst). 2010. PMID: 20573551 Free PMC article.
-
Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair.Nucleic Acids Res. 2006 Mar 23;34(6):1685-91. doi: 10.1093/nar/gkl108. Print 2006. Nucleic Acids Res. 2006. PMID: 16556909 Free PMC article.
-
Inhibition of poly(ADP-ribose) polymerase (PARP) and ataxia telangiectasia mutated (ATM) on the chemosensitivity of mantle cell lymphoma to agents that induce DNA strand breaks.Hematol Oncol. 2012 Dec;30(4):175-9. doi: 10.1002/hon.1020. Epub 2011 Dec 14. Hematol Oncol. 2012. PMID: 22170260
-
Poly(ADP-ribose) polymerase inhibition as a model for synthetic lethality in developing radiation oncology targets.Semin Radiat Oncol. 2010 Oct;20(4):274-81. doi: 10.1016/j.semradonc.2010.06.001. Semin Radiat Oncol. 2010. PMID: 20832020 Review.
-
The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax.DNA Repair (Amst). 2010 Dec 10;9(12):1273-82. doi: 10.1016/j.dnarep.2010.09.013. Epub 2010 Oct 30. DNA Repair (Amst). 2010. PMID: 21036673 Review.
Cited by
-
Tumor Cell Recovery from Senescence Induced by Radiation with PARP Inhibition.Radiat Res. 2016 Oct;186(4):327-332. doi: 10.1667/RR14437.1. Epub 2016 Sep 2. Radiat Res. 2016. PMID: 27588595 Free PMC article. Review.
-
ESO-ESMO 2nd international consensus guidelines for advanced breast cancer (ABC2)†.Ann Oncol. 2014 Oct;25(10):1871-1888. doi: 10.1093/annonc/mdu385. Epub 2014 Sep 18. Ann Oncol. 2014. PMID: 25234545 Free PMC article. No abstract available.
-
Acetylation of Werner syndrome protein (WRN): relationships with DNA damage, DNA replication and DNA metabolic activities.Biogerontology. 2014 Aug;15(4):347-66. doi: 10.1007/s10522-014-9506-3. Epub 2014 Jun 26. Biogerontology. 2014. PMID: 24965941 Free PMC article.
-
Targeting DNA-PKcs and telomerase in brain tumour cells.Mol Cancer. 2014 Oct 13;13:232. doi: 10.1186/1476-4598-13-232. Mol Cancer. 2014. PMID: 25307264 Free PMC article.
-
Case Report: Two cases of chemotherapy refractory aggressive variant prostate cancer with extreme durable response to PARP inhibitor.Front Oncol. 2025 Apr 24;15:1533627. doi: 10.3389/fonc.2025.1533627. eCollection 2025. Front Oncol. 2025. PMID: 40342821 Free PMC article.
References
-
- Goodhead DT. Initial events in the cellular effects of ionizing radiations: clustered damage in DNA. Int J Radiat Biol. 1994;65:7–17. - PubMed
-
- Radford IR. The level of induced DNA double-strand breakage correlates with cell killing after X-irradiation. Int J Radiat Biol Relat Stud Phys Chem Med. 1985;48:45–54. - PubMed
-
- Martin SA, Lord CJ, Ashworth A. DNA repair deficiency as a therapeutic target in cancer. Curr Opin Genet Dev. 2008;18:80–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
