

Next-generation genomics: an integrative approach
- PMID: 20531367
- PMCID: PMC3321268
- DOI: 10.1038/nrg2795
Next-generation genomics: an integrative approach
Abstract
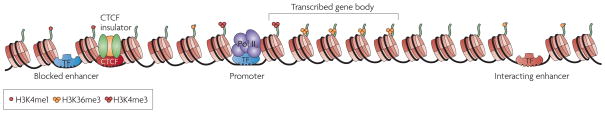
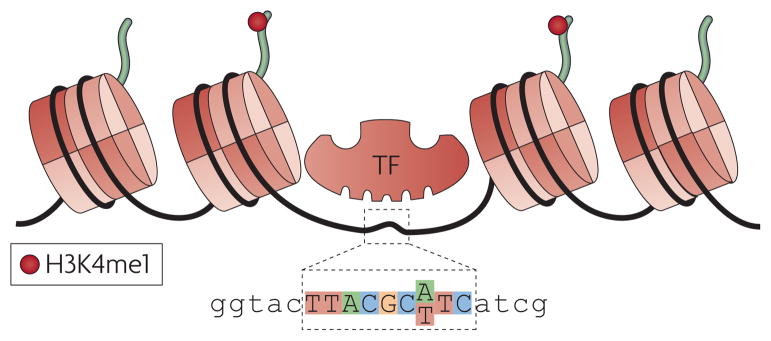
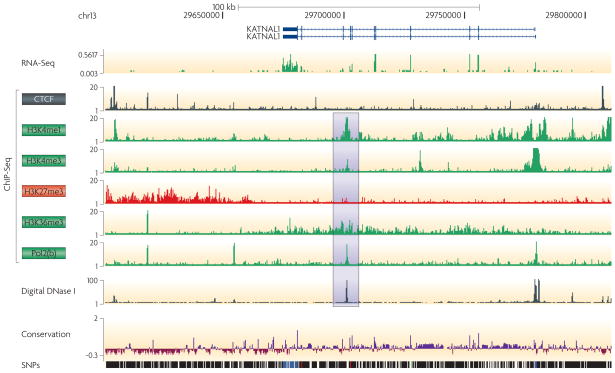
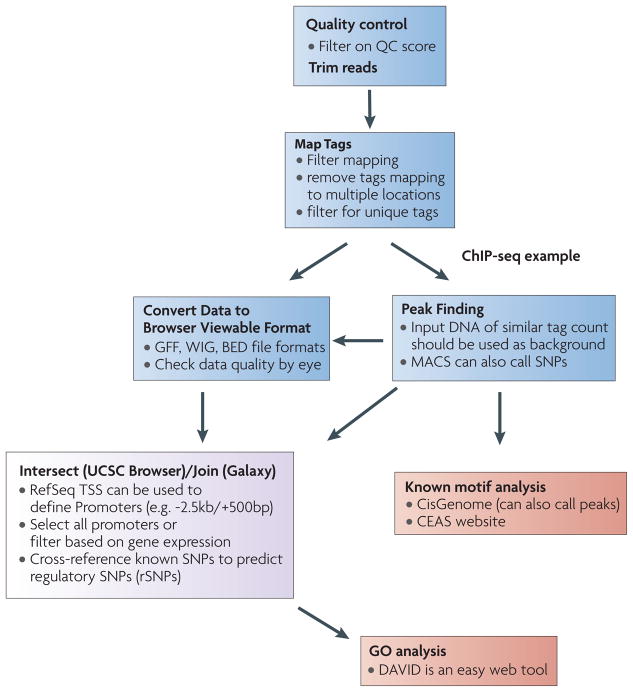
Integrating results from diverse experiments is an essential process in our effort to understand the logic of complex systems, such as development, homeostasis and responses to the environment. With the advent of high-throughput methods--including genome-wide association (GWA) studies, chromatin immunoprecipitation followed by sequencing (ChIP-seq) and RNA sequencing (RNA-seq)--acquisition of genome-scale data has never been easier. Epigenomics, transcriptomics, proteomics and genomics each provide an insightful, and yet one-dimensional, view of genome function; integrative analysis promises a unified, global view. However, the large amount of information and diverse technology platforms pose multiple challenges for data access and processing. This Review discusses emerging issues and strategies related to data integration in the era of next-generation genomics.
Figures
References
-
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
