

Endothelium-derived vasoactive factors and hypertension: possible roles in pathogenesis and as treatment targets
- PMID: 20532699
- PMCID: PMC2910890
- DOI: 10.1007/s11906-010-0118-2
Endothelium-derived vasoactive factors and hypertension: possible roles in pathogenesis and as treatment targets
Abstract
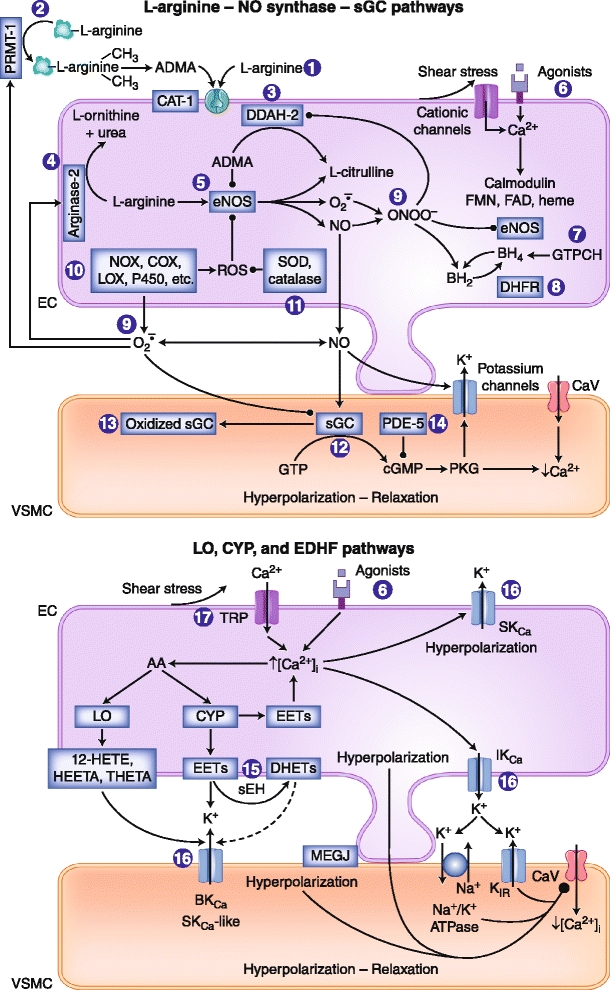
Endothelial cells regulate vascular tone by releasing various contracting and relaxing factors including nitric oxide (NO), arachidonic acid metabolites (derived from cyclooxygenases, lipoxygenases, and cytochrome P450 monooxygenases), reactive oxygen species, and vasoactive peptides. Additionally, another pathway associated with the hyperpolarization of the underlying smooth muscle cells plays a predominant role in resistance arteries. Endothelial dysfunction is a multifaceted disorder, which has been associated with hypertension of diverse etiologies, involving not only alterations of the L-arginine NO-synthase-soluble guanylyl cyclase pathway but also reduced endothelium-dependent hyperpolarizations and enhanced production of contracting factors, particularly vasoconstrictor prostanoids. This brief review highlights these different endothelial pathways as potential drug targets for novel treatments in hypertension and the associated endothelial dysfunction and end-organ damage.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
