

Going up in flames: necrotic cell injury and inflammatory diseases
- PMID: 20532807
- PMCID: PMC3051829
- DOI: 10.1007/s00018-010-0413-8
Going up in flames: necrotic cell injury and inflammatory diseases
Abstract
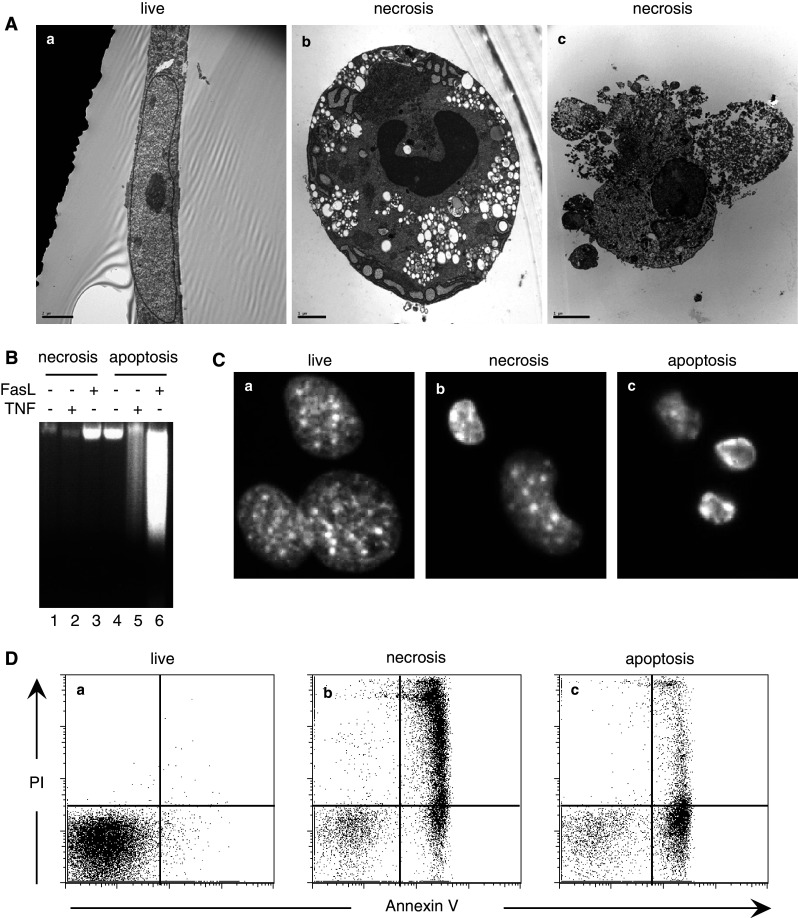
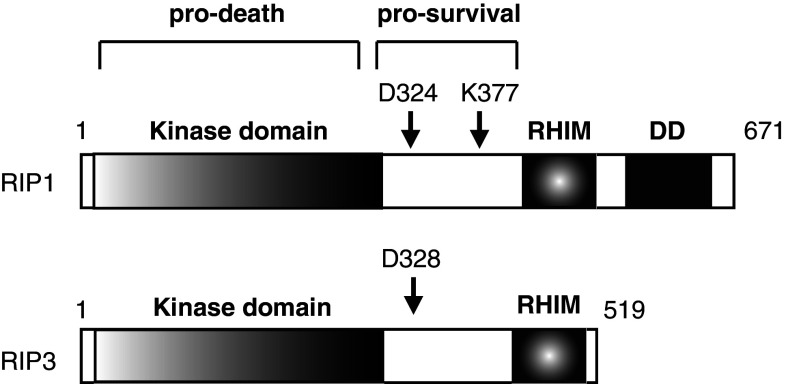
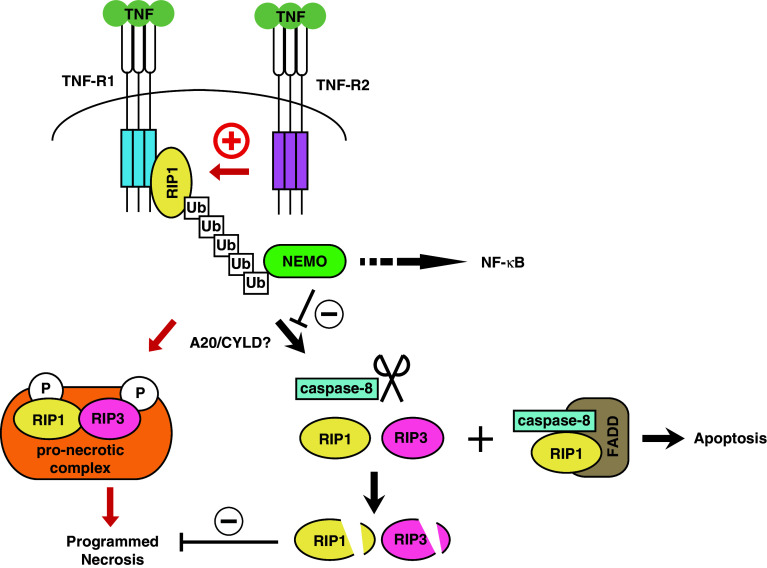
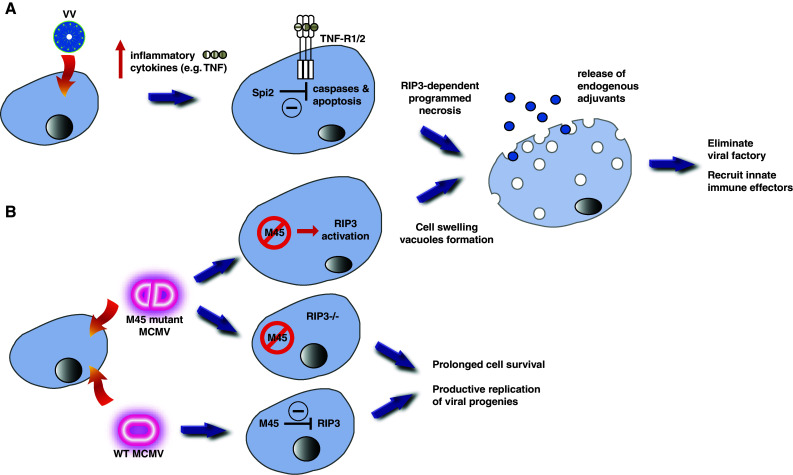
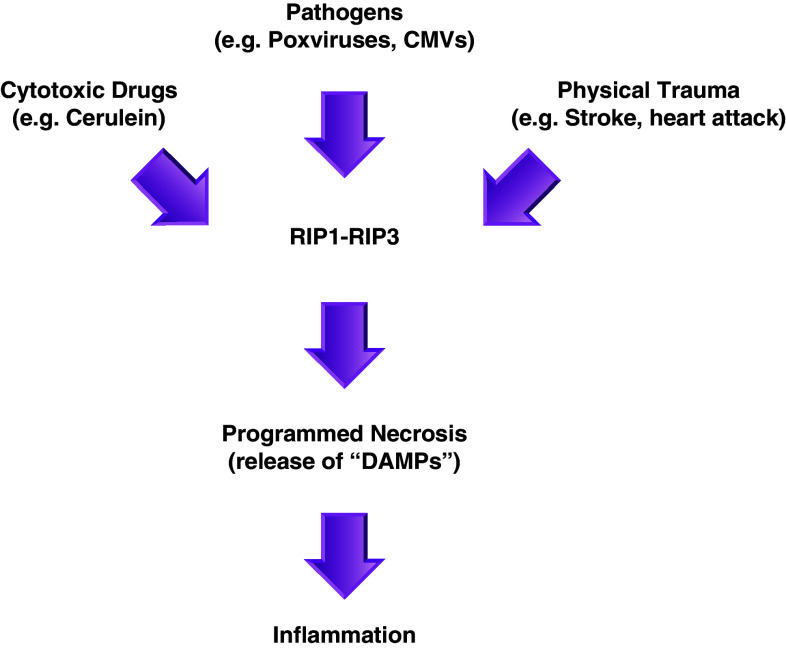
Recent evidence indicates that cell death can be induced through multiple mechanisms. Strikingly, the same death signal can often induce apoptotic as well as non-apoptotic cell death. For instance, inhibition of caspases often converts an apoptotic stimulus to one that causes necrosis. Because a dedicated molecular circuitry distinct from that controlling apoptosis is required for necrotic cell injury, terms such as "programmed necrosis" or "necroptosis" have been used to distinguish stimulus-dependent necrosis from those induced by non-specific traumas (e.g., heat shock) or secondary necrosis induced as a consequence of apoptosis. In several experimental models, programmed necrosis/necroptosis has been shown to be a crucial control point for pathogen- or injury-induced inflammation. In this review, we will discuss the molecular mechanisms that regulate programmed necrosis/necroptosis and its biological significance in pathogen infections, drug-induced cell injury, and trauma-induced tissue damage.
Figures
Similar articles
-
The molecular regulation of programmed necrotic cell injury.Trends Biochem Sci. 2010 Aug;35(8):434-41. doi: 10.1016/j.tibs.2010.03.001. Epub 2010 Mar 26. Trends Biochem Sci. 2010. PMID: 20346680 Free PMC article. Review.
-
Inflammatory outcomes of apoptosis, necrosis and necroptosis.Biol Chem. 2014 Oct;395(10):1163-71. doi: 10.1515/hsz-2014-0164. Biol Chem. 2014. PMID: 25153241 Review.
-
Necroptosis: biochemical, physiological and pathological aspects.Pathol Oncol Res. 2011 Dec;17(4):791-800. doi: 10.1007/s12253-011-9433-4. Epub 2011 Jul 21. Pathol Oncol Res. 2011. PMID: 21773880 Review.
-
Caspase-independent programmed cell death with necrotic morphology.Cell Death Differ. 1999 Jun;6(6):508-15. doi: 10.1038/sj.cdd.4400526. Cell Death Differ. 1999. PMID: 10381653 Review.
-
Is cisplatin-induced cell death always produced by apoptosis?Mol Pharmacol. 2001 Apr;59(4):657-63. doi: 10.1124/mol.59.4.657. Mol Pharmacol. 2001. PMID: 11259608 Review.
Cited by
-
Monitoring the clearance of apoptotic and necrotic cells in the nematode Caenorhabditis elegans.Methods Mol Biol. 2013;1004:183-202. doi: 10.1007/978-1-62703-383-1_14. Methods Mol Biol. 2013. PMID: 23733578 Free PMC article.
-
Dichotomy between RIP1- and RIP3-mediated necroptosis in tumor necrosis factor-α-induced shock.Mol Med. 2012 May 9;18(1):577-86. doi: 10.2119/molmed.2011.00423. Mol Med. 2012. PMID: 22371307 Free PMC article.
-
Caspase-8 Regulates Endoplasmic Reticulum Stress-Induced Necroptosis Independent of the Apoptosis Pathway in Auditory Cells.Int J Mol Sci. 2019 Nov 24;20(23):5896. doi: 10.3390/ijms20235896. Int J Mol Sci. 2019. PMID: 31771290 Free PMC article.
-
The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice.J Am Soc Nephrol. 2013 Oct;24(10):1545-57. doi: 10.1681/ASN.2012121169. Epub 2013 Jul 5. J Am Soc Nephrol. 2013. PMID: 23833261 Free PMC article.
-
How are necrotic cells recognized by their predators?Worm. 2015 Nov 30;5(1):e1120400. doi: 10.1080/21624054.2015.1120400. eCollection 2016 Jan-Mar. Worm. 2015. PMID: 27073733 Free PMC article.
References
-
- Schneider D, Gerhardt E, Bock J, Muller MM, Wolburg H, Lang F, Schulz JB. Intracellular acidification by inhibition of the Na+/H+-exchanger leads to caspase-independent death of cerebellar granule neurons resembling paraptosis. Cell Death Differ. 2004;11:760–770. doi: 10.1038/sj.cdd.4401377. - DOI - PubMed
-
- Jadus MR, Chen Y, Boldaji MT, Delgado C, Sanchez R, Douglass T, Al-Atar U, Schulz W, Lloyd C, Wepsic HT. Human U251MG glioma cells expressing the membrane form of macrophage colony-stimulating factor (mM-CSF) are killed by human monocytes in vitro and are rejected within immunodeficient mice via paraptosis that is associated with increased expression of three different heat shock proteins. Cancer Gene Ther. 2003;10:411–420. doi: 10.1038/sj.cgt.7700583. - DOI - PubMed
-
- Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J, Alnemri ES. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14:1590–1604. doi: 10.1038/sj.cdd.4402194. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
