

Synapse loss in dementias
- PMID: 20533377
- PMCID: PMC3068914
- DOI: 10.1002/jnr.22392
Synapse loss in dementias
Abstract
Synaptic transmission is essential for nervous system function, and its dysfunction is a known major contributing factor to Alzheimer's-type dementia. Antigen-specific immunochemical methods are able to characterize synapse loss in dementia through the quantification of various synaptic proteins involved in the synaptic cycle. These immunochemical methods applied to the study of Alzheimer's disease (AD) brain specimens have correlated synaptic loss with particularly toxic forms of amyloid-beta protein and have also established synapse loss as the best correlate of dementia severity. A significant but comparatively circumscribed amount of literature describes synaptic decline in other forms of dementia. Ischemic vascular dementia (IVD) is quite heterogeneous, and synapse loss in IVD seems to be variable among IVD subtypes, probably reflecting its variable neuropathologic correlates. Loss of synaptic protein has been identified in vascular dementia of the Binswanger type and Spatz-Lindenberg's disease. Here we demonstrate a significant loss of synaptophysin density within the temporal lobe of frontotemporal dementia (FTD) patients.
Figures
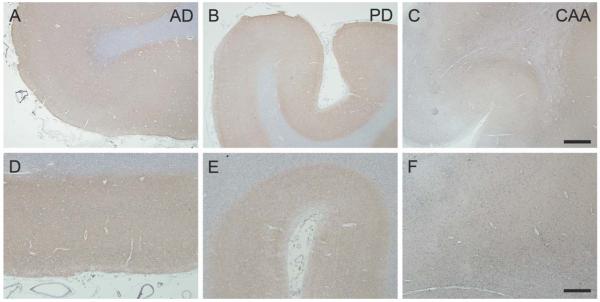
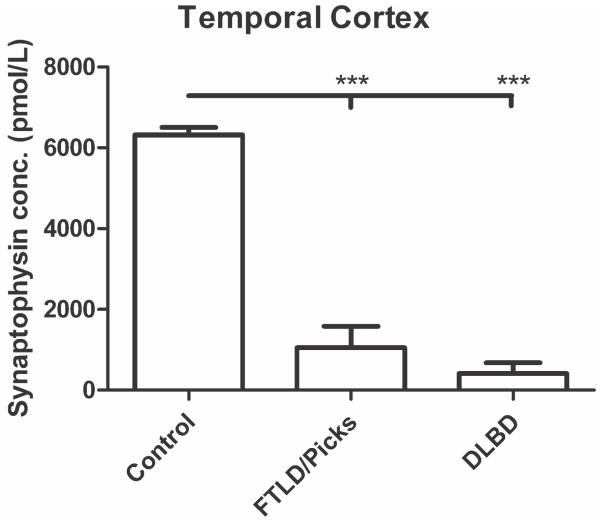
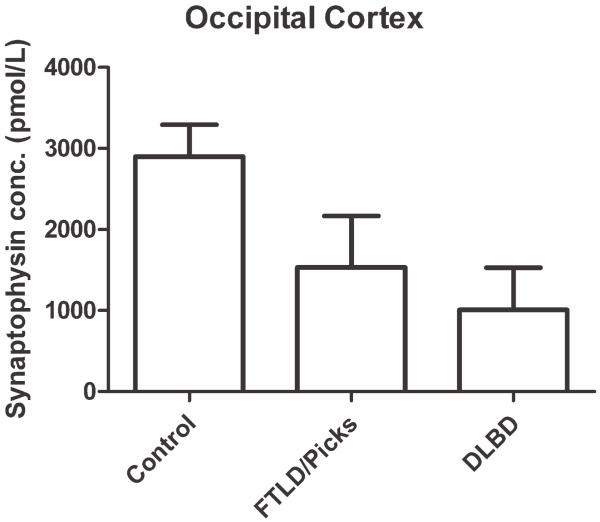
References
-
- Bergeron C, Ranalli PJ, Miceli PN. Amyloid angiopathy in Alzheimer’s disease. Can J Neurol Sci. 1987;14:564–569. - PubMed
-
- Blennow K, Bogdanovic N, Alafuzoff I, Ekman R, Davidsson P. Synaptic pathology in Alzheimer’s disease: relation to severity of dementia, but not to senile plaques, neurofibrillary tangles, or the ApoE4 allele. J Neural Transm. 1996;103:603–618. - PubMed
-
- Brown DF, Risser RC, Bigio EH, Tripp P, Stiegler A, Welch E, Eagan KP, Hladik CL, White CL., III Neocortical synapse density and Braak stage in the lewy body variant of Alzheimer disease: a comparison with classic Alzheimer disease and normal aging. J Neuropathol Exp Neurol. 1998;57:955–960. - PubMed
-
- Brun A, Liu X, Erikson C. Synapse loss and gliosis in the molecular layer of the cerebral cortex in Alzheimer’s disease and in frontal lobe degeneration. Neurodegeneration. 1995;4:171–177. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
