

Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome
- PMID: 20534480
- PMCID: PMC2890717
- DOI: 10.1073/pnas.1006214107
Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome
Abstract
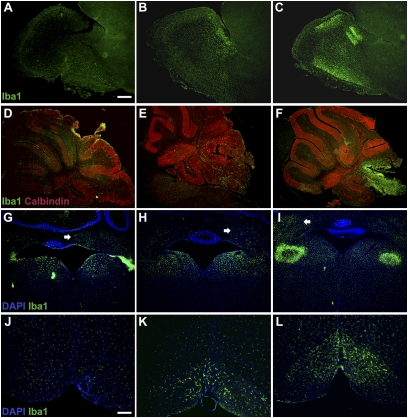
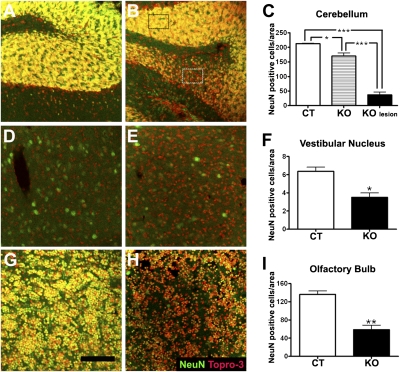
To explore the lethal, ataxic phenotype of complex I deficiency in Ndufs4 knockout (KO) mice, we inactivated Ndufs4 selectively in neurons and glia (NesKO mice). NesKO mice manifested the same symptoms as KO mice including retarded growth, loss of motor ability, breathing abnormalities, and death by approximately 7 wk. Progressive neuronal deterioration and gliosis in specific brain areas corresponded to behavioral changes as the disease advanced, with early involvement of the olfactory bulb, cerebellum, and vestibular nuclei. Neurons, particularly in these brain regions, had aberrant mitochondrial morphology. Activation of caspase 8, but not caspase 9, in affected brain regions implicate the initiation of the extrinsic apoptotic pathway. Limited caspase 3 activation and the predominance of ultrastructural features of necrotic cell death suggest a switch from apoptosis to necrosis in affected neurons. These data suggest that dysfunctional complex I in specific brain regions results in progressive glial activation that promotes neuronal death that ultimately results in mortality.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
-
- Rahman S, et al. Leigh syndrome: Clinical features and biochemical and DNA abnormalities. Ann Neurol. 1996;39:343–351. - PubMed
-
- Munaro M, et al. A single cell complementation class is common to several cases of cytochrome c oxidase-defective Leigh's syndrome. Hum Mol Genet. 1997;6:221–228. - PubMed
-
- Piao YS, Tang GC, Yang H, Lu DH. Clinico-neuropathological study of a Chinese case of familial adult Leigh syndrome. Neuropathology. 2006;26:218–221. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
