

Enhanced hepatitis C virus genome replication and lipid accumulation mediated by inhibition of AMP-activated protein kinase
- PMID: 20534540
- PMCID: PMC2895084
- DOI: 10.1073/pnas.0912426107
Enhanced hepatitis C virus genome replication and lipid accumulation mediated by inhibition of AMP-activated protein kinase
Abstract
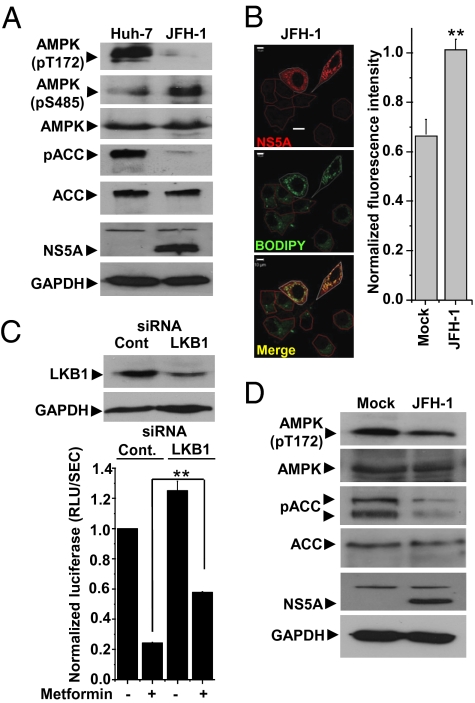
Hepatitis C virus (HCV) infection is associated with dysregulation of both lipid and glucose metabolism. As well as contributing to viral replication, these perturbations influence the pathogenesis associated with the virus, including steatosis, insulin resistance, and type 2 diabetes. AMP-activated protein kinase (AMPK) plays a key role in regulation of both lipid and glucose metabolism. We show here that, in cells either infected with HCV or harboring an HCV subgenomic replicon, phosphorylation of AMPK at threonine 172 and concomitant AMPK activity are dramatically reduced. We demonstrate that this effect is mediated by activation of the serine/threonine kinase, protein kinase B, which inhibits AMPK by phosphorylating serine 485. The physiological significance of this inhibition is demonstrated by the observation that pharmacological restoration of AMPK activity not only abrogates the lipid accumulation observed in virus-infected and subgenomic replicon-harboring cells but also efficiently inhibits viral replication. These data demonstrate that inhibition of AMPK is required for HCV replication and that the restoration of AMPK activity may present a target for much needed anti-HCV therapies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558–567. - PubMed
-
- Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5:453–463. - PubMed
-
- McLauchlan J. Lipid droplets and hepatitis C virus infection. Biochim Biophys Acta. 2009;1791:552–559. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
