

Activity of any class IA PI3K isoform can sustain cell proliferation and survival
- PMID: 20534549
- PMCID: PMC2895061
- DOI: 10.1073/pnas.0906461107
Activity of any class IA PI3K isoform can sustain cell proliferation and survival
Abstract
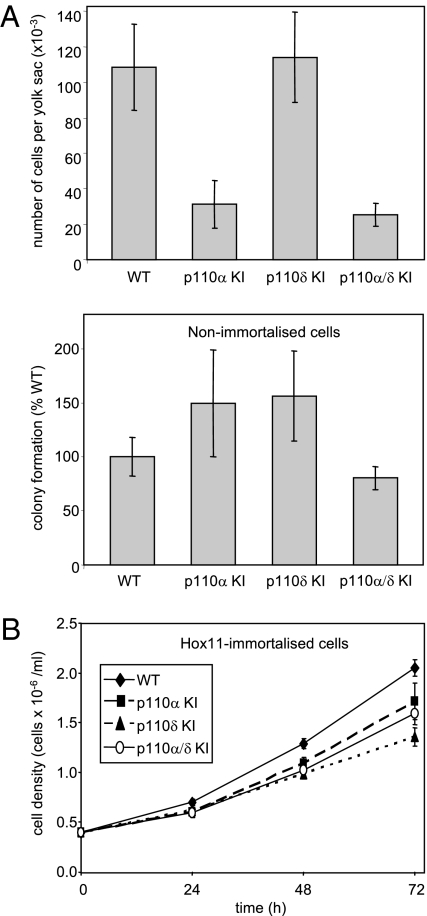
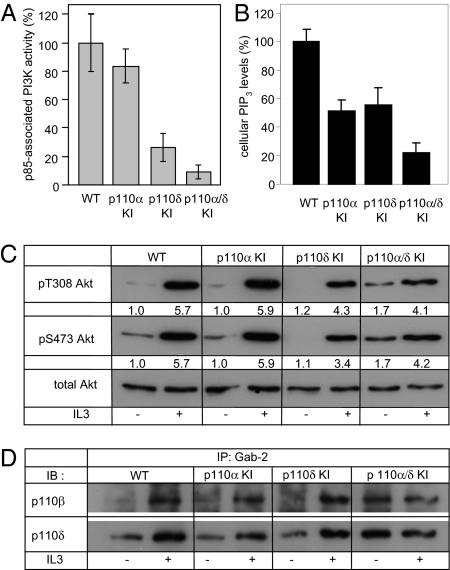
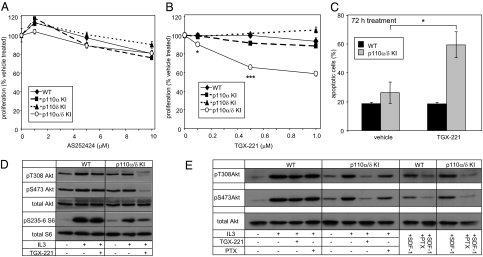
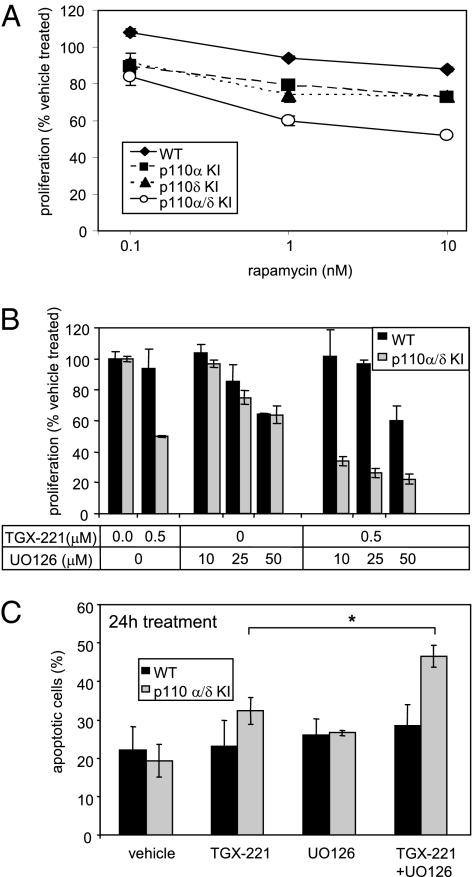
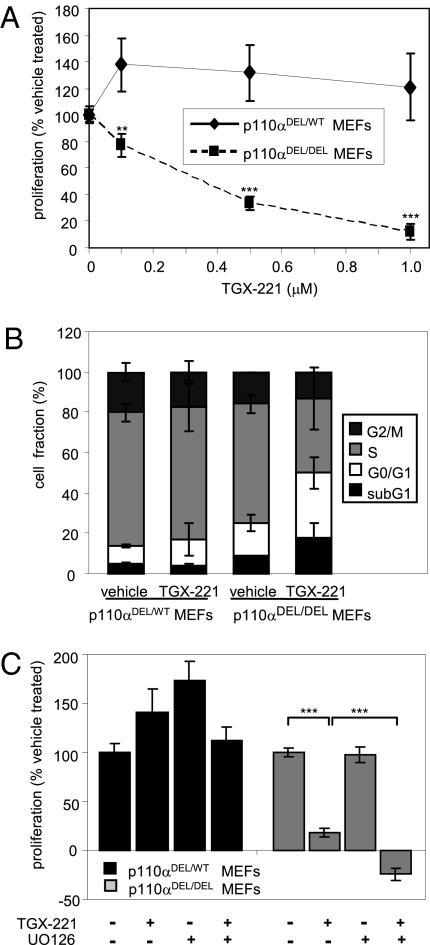
Small molecule inhibitors of PI3K for oncology mainly target the class I PI3Ks, comprising the p110alpha, beta, gamma, and delta isoforms, of which only p110alpha is mutated in cancer. To assess the roles of class I PI3K isoforms in cell proliferation and survival, we generated immortalized mouse leukocyte and fibroblast models in which class I PI3Ks were inactivated by genetic and pharmacological approaches. In IL3-dependent hemopoietic progenitor cells (which express all four class I PI3K isoforms), genetic inactivation of either p110alpha or p110delta did not affect cell proliferation or survival or sensitize to p110beta or p110gamma inactivation. Upon compound inactivation of p110alpha and p110delta, which removed >90% of p85-associated PI3K activity, remarkably, cells continued to proliferate effectively, with p110beta assuming an essential role in signaling and cell survival. Furthermore, under these conditions of diminished class I PI3K activity, input from the ERK pathway became important for cell survival. Similar observations were made in mouse embryonic fibroblasts (which mainly express p110alpha and p110beta) in which p110alpha or p110beta could sustain cell proliferation as a single isoform. Taken together, these data demonstrate that a small fraction of total class I PI3K activity is sufficient to sustain cell survival and proliferation. Persistent inhibition of selected PI3K isoforms can allow the remaining isoform(s) to couple to upstream signaling pathways in which they are not normally engaged. Such functional redundancy of class IA PI3K isoforms upon sustained PI3K inhibition has implications for the development and use of PI3K inhibitors in cancer.
Conflict of interest statement
Conflict of interest statement: Bart Vanhaesebroeck is a consultant for Intellikine (San Diego, CA).
Figures
References
-
- Kok K, Geering B, Vanhaesebroeck B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem Sci. 2009;34:115–127. - PubMed
-
- Shayesteh L, et al. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet. 1999;21:99–102. - PubMed
-
- Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. - PubMed
-
- Vogt PK, Kang S, Elsliger MA, Gymnopoulos M. Cancer-specific mutations in phosphatidylinositol 3-kinase. Trends Biochem Sci. 2007;32:342–349. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
