

An application of supertree methods to Mammalian mitogenomic sequences
- PMID: 20535231
- PMCID: PMC2880846
- DOI: 10.4137/ebo.s4527
An application of supertree methods to Mammalian mitogenomic sequences
Abstract
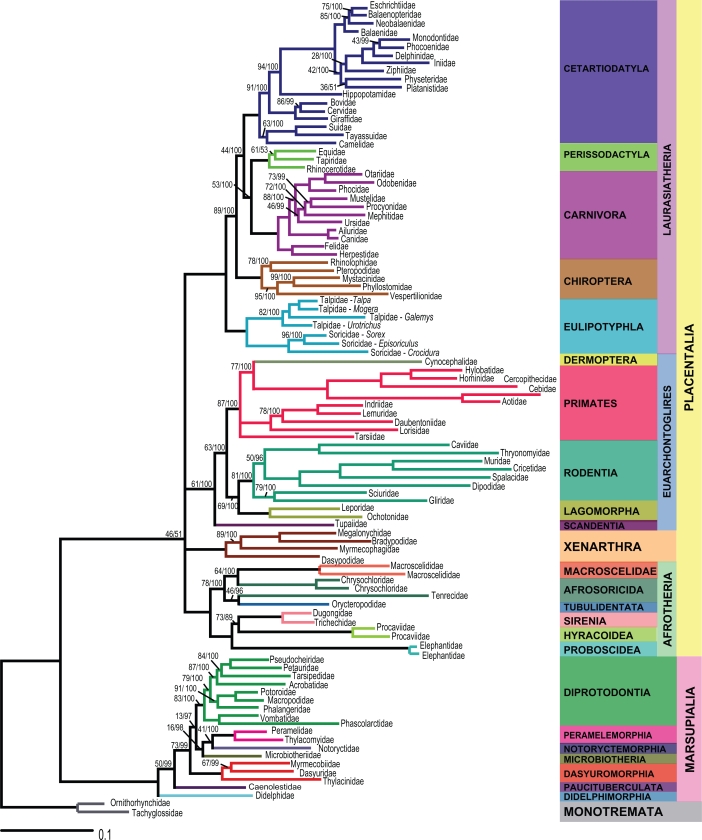
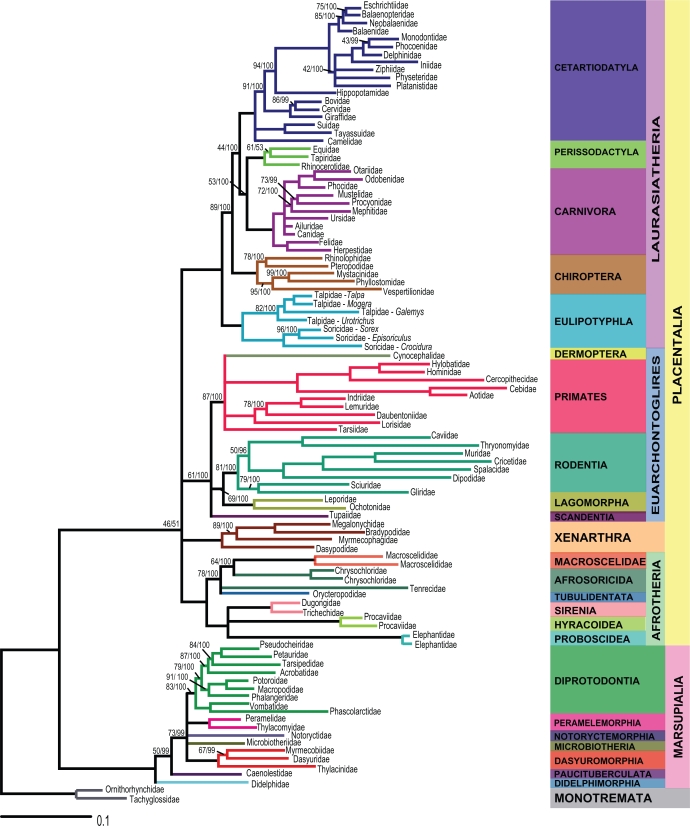
TWO DIFFERENT APPROACHES CAN BE USED IN PHYLOGENOMICS: combined or separate analysis. In the first approach, different datasets are combined in a concatenated supermatrix. In the second, datasets are analyzed separately and the phylogenetic trees are then combined in a supertree. The supertree method is an interesting alternative to avoid missing data, since datasets that are analyzed separately do not need to represent identical taxa. However, the supertree approach and the corresponding consensus methods have been highly criticized for not providing valid phylogenetic hypotheses. In this study, congruence of trees estimated by consensus and supertree approaches were compared to model trees obtained from a combined analysis of complete mitochondrial sequences of 102 species representing 93 mammal families. The consensus methods produced poorly resolved consensus trees and did not perform well, except for the majority rule consensus with compatible groupings. The weighted supertree and matrix representation with parsimony methods performed equally well and were highly congruent with the model trees. The most similar supertree method was the least congruent with the model trees. We conclude that some of the methods tested are worth considering in a phylogenomic context.
Keywords: DNA sequences; combined analysis; consensus; phylogenomics; separate analysis; supermatrix.
Figures
Similar articles
-
Complete generic-level phylogenetic analyses of palms (Arecaceae) with comparisons of supertree and supermatrix approaches.Syst Biol. 2009 Apr;58(2):240-56. doi: 10.1093/sysbio/syp021. Epub 2009 May 30. Syst Biol. 2009. PMID: 20525581
-
A simulation study comparing supertree and combined analysis methods using SMIDGen.Algorithms Mol Biol. 2010 Jan 4;5:8. doi: 10.1186/1748-7188-5-8. Algorithms Mol Biol. 2010. PMID: 20047664 Free PMC article.
-
A comparison of supermatrix and supertree methods for multilocus phylogenetics using organismal datasets.Cladistics. 2013 Oct;29(5):560-566. doi: 10.1111/cla.12014. Epub 2013 Feb 18. Cladistics. 2013. PMID: 34798766
-
Genus-level supertree of Cyprinidae (Actinopterygii: Cypriniformes), partitioned qualitative clade support and test of macro-evolutionary scenarios.Biol Rev Camb Philos Soc. 2009 Nov;84(4):653-89. doi: 10.1111/j.1469-185X.2009.00091.x. Biol Rev Camb Philos Soc. 2009. PMID: 19857213 Review.
-
Molecular and morphological supertree of stony corals (Anthozoa: Scleractinia) using matrix representation parsimony.Biol Rev Camb Philos Soc. 2005 Nov;80(4):543-58. doi: 10.1017/S1464793105006780. Biol Rev Camb Philos Soc. 2005. PMID: 16221328 Review.
Cited by
-
Diversification and the rate of molecular evolution: no evidence of a link in mammals.BMC Evol Biol. 2011 Oct 4;11:286. doi: 10.1186/1471-2148-11-286. BMC Evol Biol. 2011. PMID: 21967038 Free PMC article.
-
Controversies in modern evolutionary biology: the imperative for error detection and quality control.BMC Genomics. 2012 Jan 4;13:5. doi: 10.1186/1471-2164-13-5. BMC Genomics. 2012. PMID: 22217008 Free PMC article.
References
-
- Rokas A, Williams BL, King N, Carroll SB. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 2003;425(6960):798–804. - PubMed
-
- Driskell AC, Ané C, Burleigh JG, McMahon MM, O’Meara BC, Sanderson MJ. Prospects for building the tree of life from large sequence databases. Science. 2004;306(5699):1172–4. - PubMed
-
- Rodriguez-Ezpeleta N, Brinkmann H, Burey SC, Roure B, Burger G, Loffelhardt W, et al. Monophyly of primary photosynthetic eukaryotes: Green plants, red algae, and glaucophytes. Curr Biol. 2005;15(14):1325–30. - PubMed
-
- Dunn CW, Hejnol A, Matus DQ, Pang K, Browne WE, Smith SA, et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 2008;452(7188):745–9. - PubMed
-
- Delsuc F, Brinkmann H, Philippe H. Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet. 2005;6(5):361–75. - PubMed
LinkOut - more resources
Full Text Sources
