

Comparative genomics of the family Vibrionaceae reveals the wide distribution of genes encoding virulence-associated proteins
- PMID: 20537180
- PMCID: PMC2890568
- DOI: 10.1186/1471-2164-11-369
Comparative genomics of the family Vibrionaceae reveals the wide distribution of genes encoding virulence-associated proteins
Abstract
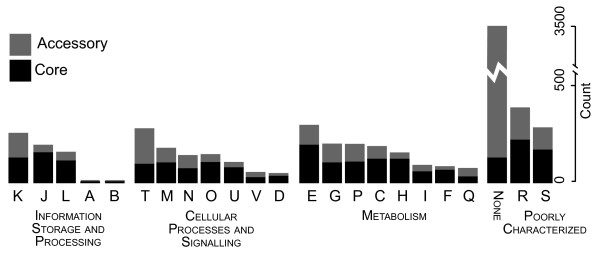
Background: Species of the family Vibrionaceae are ubiquitous in marine environments. Several of these species are important pathogens of humans and marine species. Evidence indicates that genetic exchange plays an important role in the emergence of new pathogenic strains within this family. Data from the sequenced genomes of strains in this family could show how the genes encoded by all these strains, known as the pangenome, are distributed. Information about the core, accessory and panproteome of this family can show how, for example, genes encoding virulence-associated proteins are distributed and help us understand how virulence emerges.
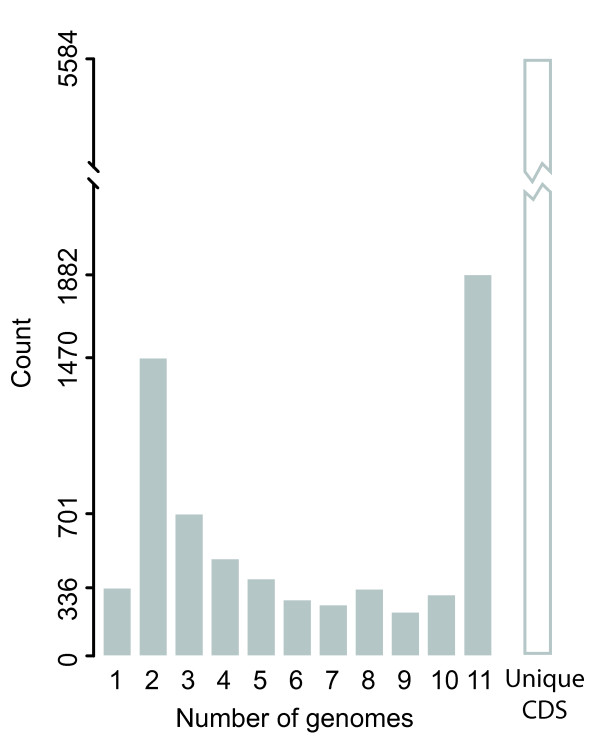
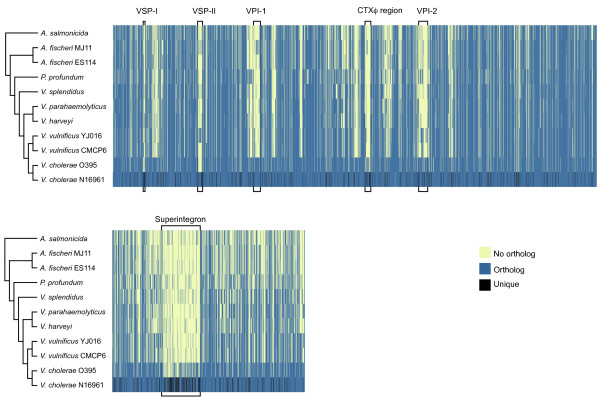
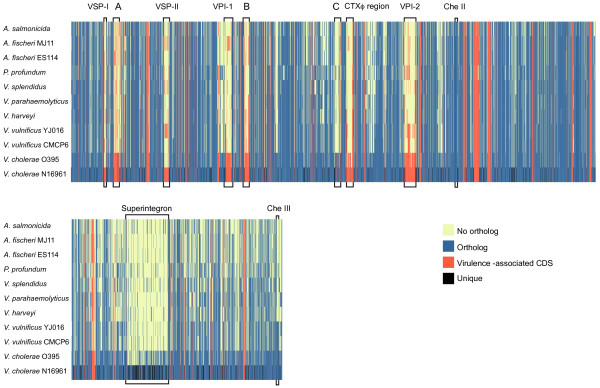
Results: We deduced the complete set of orthologs for eleven strains from this family. The core proteome consists of 1,882 orthologous groups, which is 28% of the 6,629 orthologous groups in this family. There were 4,411 accessory orthologous groups (i.e., proteins that occurred in from 2 to 10 proteomes) and 5,584 unique proteins (encoded once on only one of the eleven genomes). Proteins that have been associated with virulence in V. cholerae were widely distributed across the eleven genomes, but the majority was found only on the genomes of the two V. cholerae strains examined.
Conclusions: The proteomes are reflective of the differing evolutionary trajectories followed by different strains to similar phenotypes. The composition of the proteomes supports the notion that genetic exchange among species of the Vibrionaceae is widespread and that this exchange aids these species in adapting to their environments.
Figures
Similar articles
-
Genomic and systems evolution in Vibrionaceae species.BMC Genomics. 2009 Jul 7;10 Suppl 1(Suppl 1):S11. doi: 10.1186/1471-2164-10-S1-S11. BMC Genomics. 2009. PMID: 19594870 Free PMC article.
-
Comparative genomics of the bacterial genus Listeria: Genome evolution is characterized by limited gene acquisition and limited gene loss.BMC Genomics. 2010 Dec 2;11:688. doi: 10.1186/1471-2164-11-688. BMC Genomics. 2010. PMID: 21126366 Free PMC article.
-
Genes similar to the Vibrio parahaemolyticus virulence-related genes tdh, tlh, and vscC2 occur in other vibrionaceae species isolated from a pristine estuary.Appl Environ Microbiol. 2014 Jan;80(2):595-602. doi: 10.1128/AEM.02895-13. Epub 2013 Nov 8. Appl Environ Microbiol. 2014. PMID: 24212573 Free PMC article.
-
[The pathogenicity factors of opportunistic bacteria in the family Vibrionaceae (I)].Bacteriol Virusol Parazitol Epidemiol. 1996 Jul-Dec;41(3-4):99-105. Bacteriol Virusol Parazitol Epidemiol. 1996. PMID: 9116408 Review. Romanian. No abstract available.
-
Small RNAs in the Vibrionaceae: an ocean still to be explored.Wiley Interdiscip Rev RNA. 2014 May-Jun;5(3):381-92. doi: 10.1002/wrna.1218. Epub 2014 Jan 23. Wiley Interdiscip Rev RNA. 2014. PMID: 24458378 Review.
Cited by
-
Visual analytics of surveillance data on foodborne vibriosis, United States, 1973-2010.Environ Health Insights. 2011;5:71-85. doi: 10.4137/EHI.S7806. Epub 2011 Nov 10. Environ Health Insights. 2011. PMID: 22174586 Free PMC article.
-
The core and unique proteins of haloarchaea.BMC Genomics. 2012 Jan 24;13:39. doi: 10.1186/1471-2164-13-39. BMC Genomics. 2012. PMID: 22272718 Free PMC article.
-
Recovery and Community Succession of the Zostera marina Rhizobiome after Transplantation.Appl Environ Microbiol. 2021 Jan 15;87(3):e02326-20. doi: 10.1128/AEM.02326-20. Print 2021 Jan 15. Appl Environ Microbiol. 2021. PMID: 33187993 Free PMC article.
-
Comprehensive genomic and evolutionary analysis of biofilm matrix clusters and proteins in the Vibrio genus.mSystems. 2025 May 20;10(5):e0006025. doi: 10.1128/msystems.00060-25. Epub 2025 Apr 10. mSystems. 2025. PMID: 40207939 Free PMC article.
-
Vibrio cholerae can Recycle Fatty Acids Via an Acyl-Acyl Carrier Protein Synthetase.Curr Microbiol. 2025 Jun 30;82(8):352. doi: 10.1007/s00284-025-04332-9. Curr Microbiol. 2025. PMID: 40586929 Free PMC article.
References
-
- Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS. et al.Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial "pan-genome". Proc Natl Acad Sci USA. 2005;102(39):13950–13955. doi: 10.1073/pnas.0506758102. - DOI - PMC - PubMed
-
- Han CS, Xie G, Challacombe JF, Altherr MR, Bhotika SS, Brown N, Bruce D, Campbell CS, Campbell ML, Chen J. et al.Pathogenomic sequence analysis of Bacillus cereus and Bacillus thuringiensis isolates closely related to Bacillus anthracis. J Bacteriol. 2006;188(9):3382–3390. doi: 10.1128/JB.188.9.3382-3390.2006. - DOI - PMC - PubMed
-
- Rasko DA, Rosovitz MJ, Myers GS, Mongodin EF, Fricke WF, Gajer P, Crabtree J, Sperandio V, Ravel J. The pan-genome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol. 2008;190(20):6881–6893. doi: 10.1128/JB.00619-08. - DOI - PMC - PubMed
-
- Schoen C, Blom J, Claus H, Schramm-Glück A, Brandt P, Müller T, Goesmann A, Joseph B, Konietzny S, Kurzai O. et al.Whole-genome comparison of disease and carriage strains provides insights into virulence evolution in Neisseria meningitidis. Proc Natl Acad Sci USA. 2008;105(9):3473–3478. doi: 10.1073/pnas.0800151105. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
