

Glycogen synthase kinase 3 beta: can it be a target for oral cancer
- PMID: 20537194
- PMCID: PMC2906469
- DOI: 10.1186/1476-4598-9-144
Glycogen synthase kinase 3 beta: can it be a target for oral cancer
Abstract
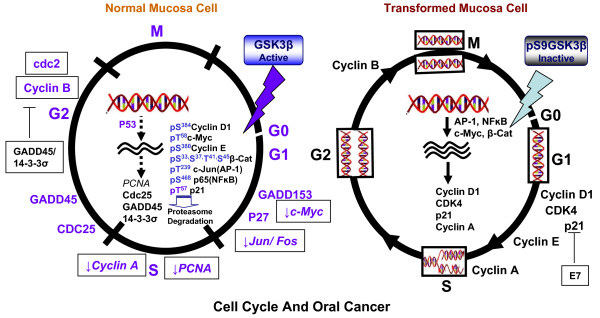
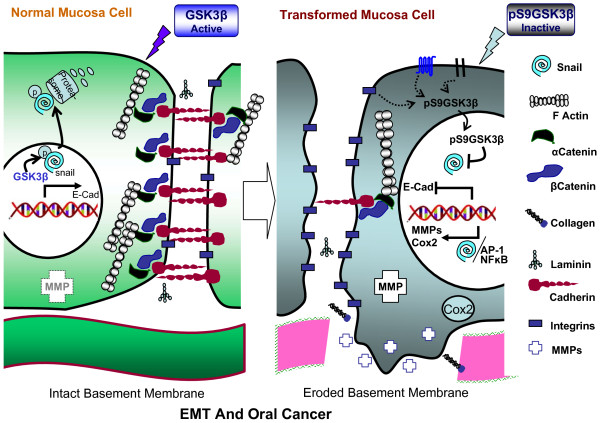
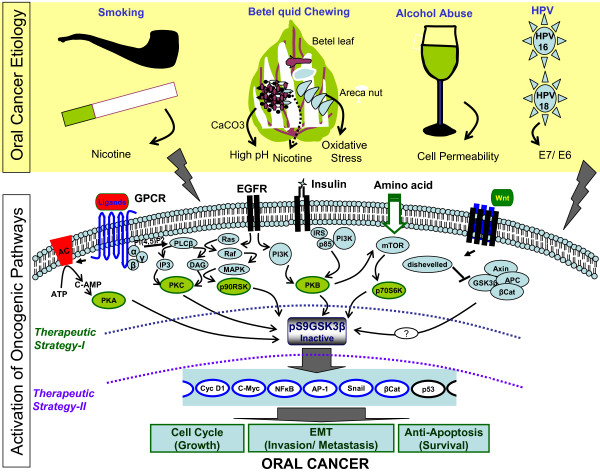
Despite progress in treatment approaches for oral cancer, there has been only modest improvement in patient outcomes in the past three decades. The frequent treatment failure is due to the failure to control tumor recurrence and metastasis. These failures suggest that new targets should be identified to reverse oral epithelial dysplastic lesions. Recent developments suggest an active role of glycogen synthase kinase 3 beta (GSK3 beta) in various human cancers either as a tumor suppressor or as a tumor promoter. GSK3beta is a Ser/Thr protein kinase, and there is emerging evidence that it is a tumor suppressor in oral cancer. The evidence suggests a link between key players in oral cancer that control transcription, accelerated cell cycle progression, activation of invasion/metastasis and anti-apoptosis, and regulation of these factors by GSK3beta. Moreover, the major upstream kinases of GSK3beta and their oncogenic activation by several etiological agents of oral cancer support this hypothesis. In spite of all this evidence, a detailed analysis of the role of GSK3beta in oral cancer and of its therapeutic potential has yet to be conducted by the scientific community. The focus of this review is to discuss the multitude of roles of GSK3beta, its possible role in controlling different oncogenic events and how it can be targeted in oral cancer.
Figures

Similar articles
-
Glycogen synthase kinase-3β mediated regulation of matrix metalloproteinase-9 and its involvement in oral squamous cell carcinoma progression and invasion.Cell Oncol (Dordr). 2018 Feb;41(1):47-60. doi: 10.1007/s13402-017-0358-0. Epub 2017 Nov 13. Cell Oncol (Dordr). 2018. PMID: 29134466
-
Reversion-inducing cysteine-rich protein with Kazal motifs and its regulation by glycogen synthase kinase 3 signaling in oral cancer.Tumour Biol. 2016 Nov;37(11):15253-15264. doi: 10.1007/s13277-016-5362-x. Epub 2016 Sep 30. Tumour Biol. 2016. PMID: 27696293
-
Expression and inactivation of glycogen synthase kinase 3 alpha/ beta and their association with the expression of cyclin D1 and p53 in oral squamous cell carcinoma progression.Mol Cancer. 2015 Feb 3;14:20. doi: 10.1186/s12943-015-0300-x. Mol Cancer. 2015. PMID: 25645517 Free PMC article.
-
An emerging strategy for cancer treatment targeting aberrant glycogen synthase kinase 3 beta.Anticancer Agents Med Chem. 2009 Dec;9(10):1114-22. doi: 10.2174/187152009789734982. Anticancer Agents Med Chem. 2009. PMID: 19925395 Review.
-
Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and cancer chemotherapy.Cancer Lett. 2009 Jan 18;273(2):194-200. doi: 10.1016/j.canlet.2008.05.045. Epub 2008 Jul 7. Cancer Lett. 2009. PMID: 18606491 Free PMC article. Review.
Cited by
-
Modulation of deoxycytidine kinase (dCK) and glycogen synthase kinase (GSK-3β) by anti-CD20 (rituximab) and 2-chlorodeoxyadenosine (2-CdA) in human lymphoid malignancies.Exp Hematol Oncol. 2014 Dec 19;3:31. doi: 10.1186/2162-3619-3-31. eCollection 2014. Exp Hematol Oncol. 2014. PMID: 25937997 Free PMC article.
-
Dietary phytochemicals and cancer prevention: Nrf2 signaling, epigenetics, and cell death mechanisms in blocking cancer initiation and progression.Pharmacol Ther. 2013 Feb;137(2):153-71. doi: 10.1016/j.pharmthera.2012.09.008. Epub 2012 Oct 3. Pharmacol Ther. 2013. PMID: 23041058 Free PMC article. Review.
-
Discrete functions of GSK3α and GSK3β isoforms in prostate tumor growth and micrometastasis.Oncotarget. 2015 Mar 20;6(8):5947-62. doi: 10.18632/oncotarget.3335. Oncotarget. 2015. PMID: 25714023 Free PMC article.
-
GSK-3α Is a Novel Target of CREB and CREB-GSK-3α Signaling Participates in Cell Viability in Lung Cancer.PLoS One. 2016 Apr 6;11(4):e0153075. doi: 10.1371/journal.pone.0153075. eCollection 2016. PLoS One. 2016. PMID: 27049759 Free PMC article.
-
[Expression and clinical significance of nuclear factor κB/B cell lymphoma-2 signal pathway and glycogen synthase kinase 3β in oral squamous cell carcinoma].Hua Xi Kou Qiang Yi Xue Za Zhi. 2015 Feb;33(1):11-5. doi: 10.7518/hxkq.2015.01.003. Hua Xi Kou Qiang Yi Xue Za Zhi. 2015. PMID: 25872291 Free PMC article. Chinese.
References
-
- Cheong SC, Chandramouli GV, Saleh A, Zain RB, Lau SH, Sivakumaren S, Pathmanathan R, Prime SS, Teo SH, Patel V, Gutkind JS. Gene expression in human oral squamous cell carcinoma is influenced by risk factor exposure. Oral Oncol. 2009;45:712–719. doi: 10.1016/j.oraloncology.2008.11.002. - DOI - PubMed
-
- Mishra A, Bharti AC, Saluja D, Das BC. Transactivation and expression patterns of Jun and Fos/AP-1 super-family proteins in human oral cancer. Int J Cancer. 2010;126:819–829. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
