

Tracing the tumor lineage
- PMID: 20537601
- PMCID: PMC2904844
- DOI: 10.1016/j.molonc.2010.04.010
Tracing the tumor lineage
Erratum in
- Mol Oncol. 2011 Jun;5(3):302
Abstract
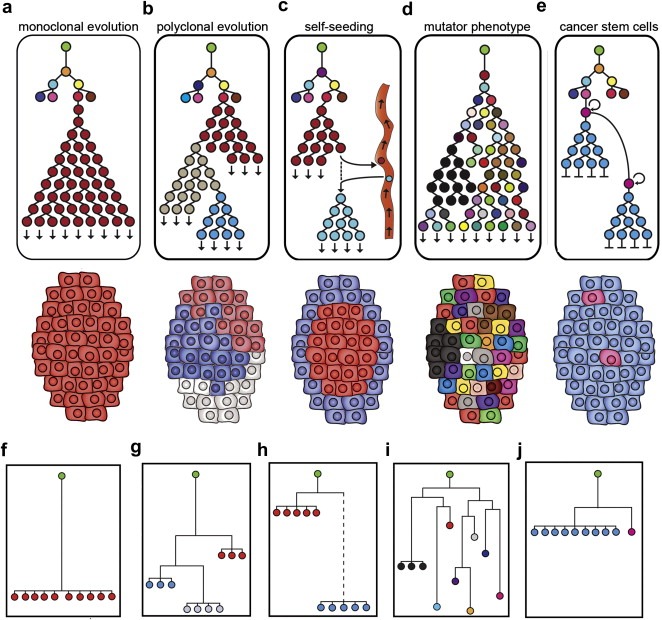
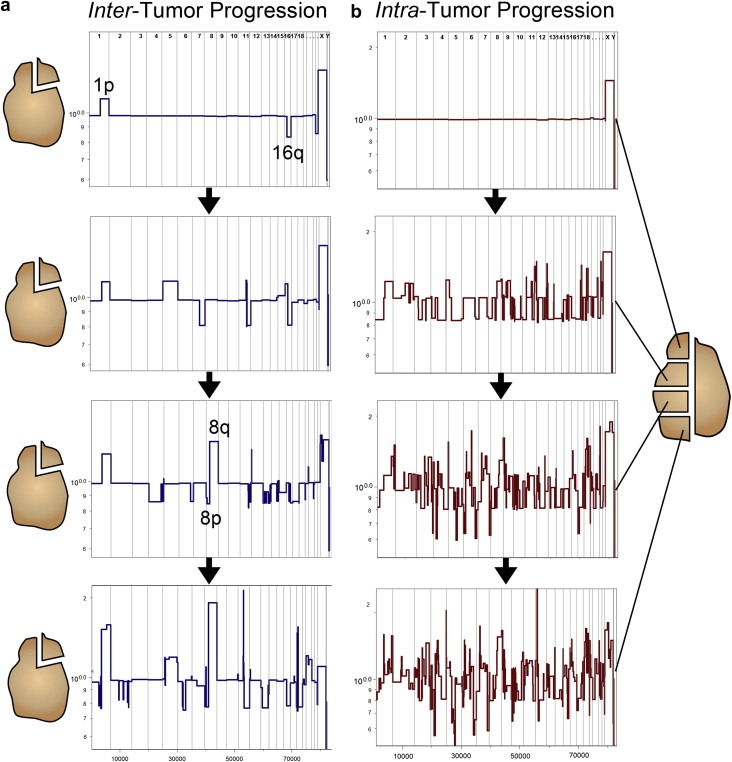
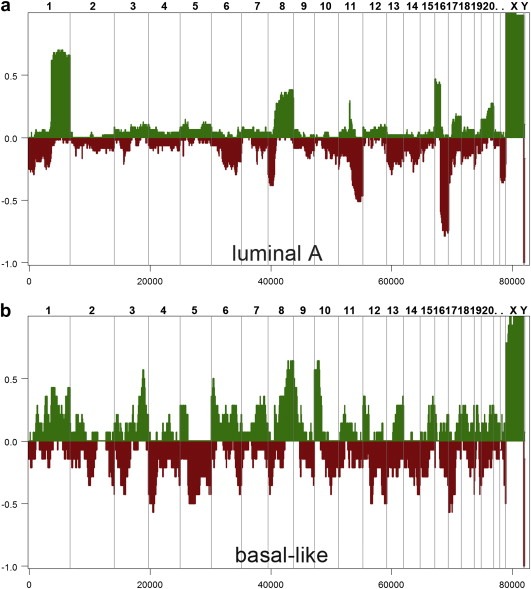
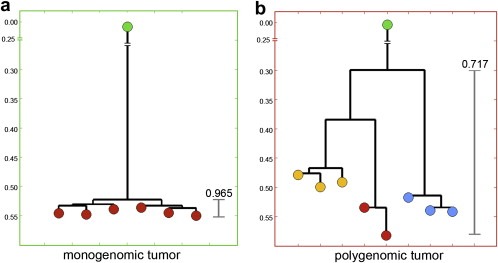
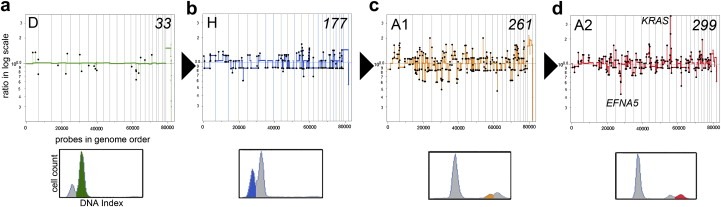
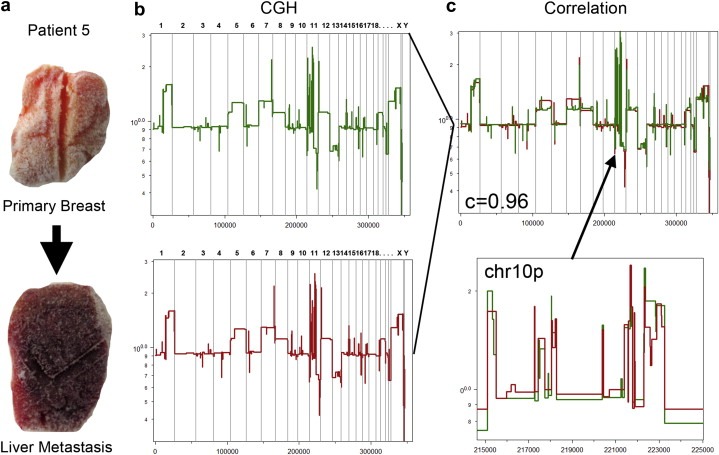
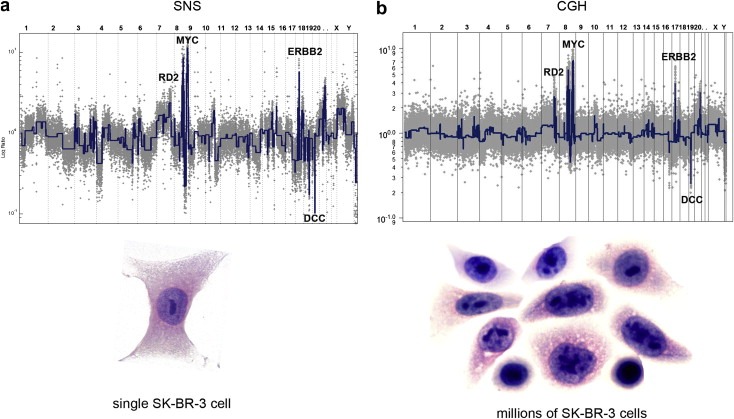
Defining the pathways through which tumors progress is critical to our understanding and treatment of cancer. We do not routinely sample patients at multiple time points during the progression of their disease, and thus our research is limited to inferring progression a posteriori from the examination of a single tumor sample. Despite this limitation, inferring progression is possible because the tumor genome contains a natural history of the mutations that occur during the formation of the tumor mass. There are two approaches to reconstructing a lineage of progression: (1) inter-tumor comparisons, and (2) intra-tumor comparisons. The inter-tumor approach consists of taking single samples from large collections of tumors and comparing the complexity of the genomes to identify early and late mutations. The intra-tumor approach involves taking multiple samples from individual heterogeneous tumors to compare divergent clones and reconstruct a phylogenetic lineage. Here we discuss how these approaches can be used to interpret the current models for tumor progression. We also compare data from primary and metastatic copy number profiles to shed light on the final steps of breast cancer progression. Finally, we discuss how recent technical advances in single cell genomics will herald a new era in understanding the fundamental basis of tumor heterogeneity and progression.
(c) 2010 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Adams, J.M. , Strasser, A. , 2008. Is tumor growth sustained by rare cancer stem cells or dominant clones?. Cancer Res. 68, 4018–4021. - PubMed
-
- Allred, D.C. , Wu, Y. , Mao, S. , Nagtegaal, I.D. , Lee, S. , Perou, C.M. , Mohsin, S.K. , O'Connell, P. , Tsimelzon, A. , Medina, D. , 2008. Ductal carcinoma in situ and the emergence of diversity during breast cancer evolution. Clin. Cancer Res. 14, 370–378. - PubMed
-
- Aubele, M. , Mattis, A. , Zitzelsberger, H. , Walch, A. , Kremer, M. , Hutzler, P. , Höfler, H. , Werner, M. , 1999. Intratumoral heterogeneity in breast carcinoma revealed by laser-microdissection and comparative genomic hybridization. Cancer Genet. Cytogenet. 110, 94–102. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
