

Autoactivation of transforming growth factor beta-activated kinase 1 is a sequential bimolecular process
- PMID: 20538596
- PMCID: PMC2919138
- DOI: 10.1074/jbc.M109.093468
Autoactivation of transforming growth factor beta-activated kinase 1 is a sequential bimolecular process
Abstract
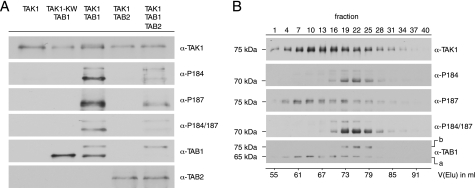
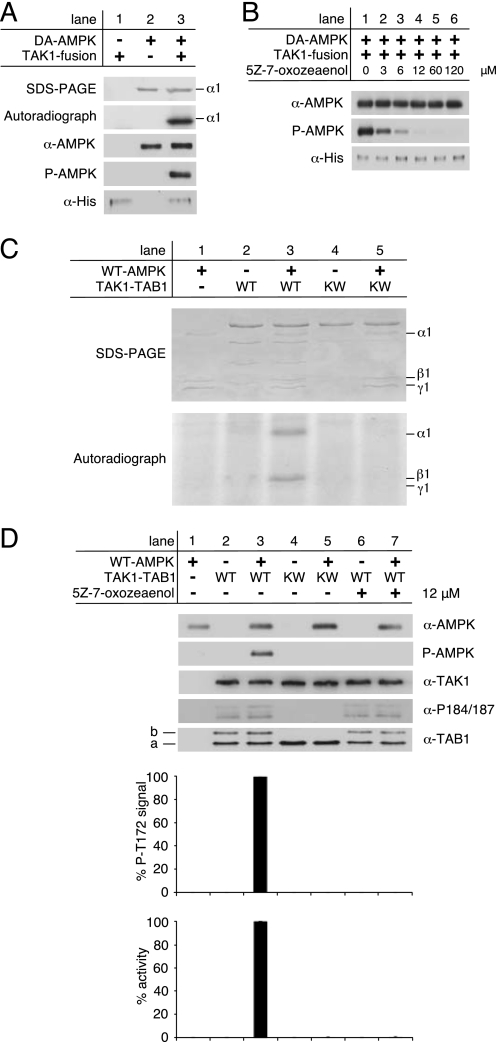
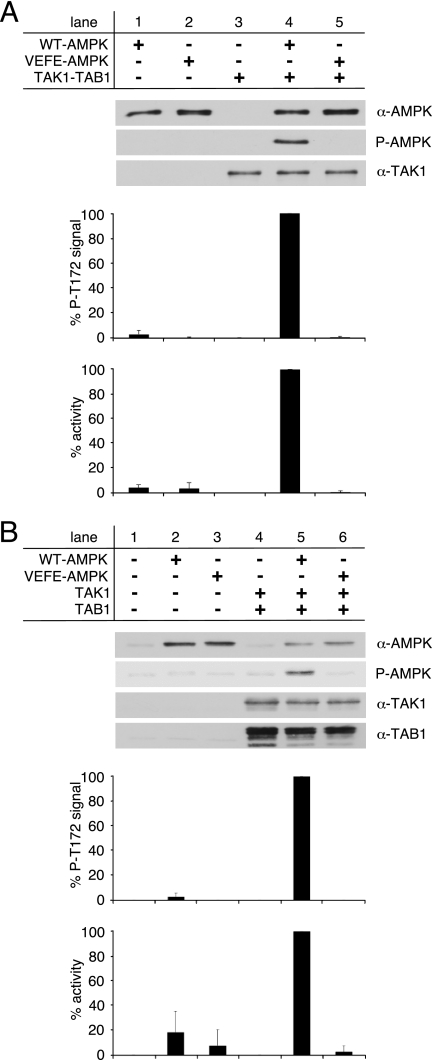
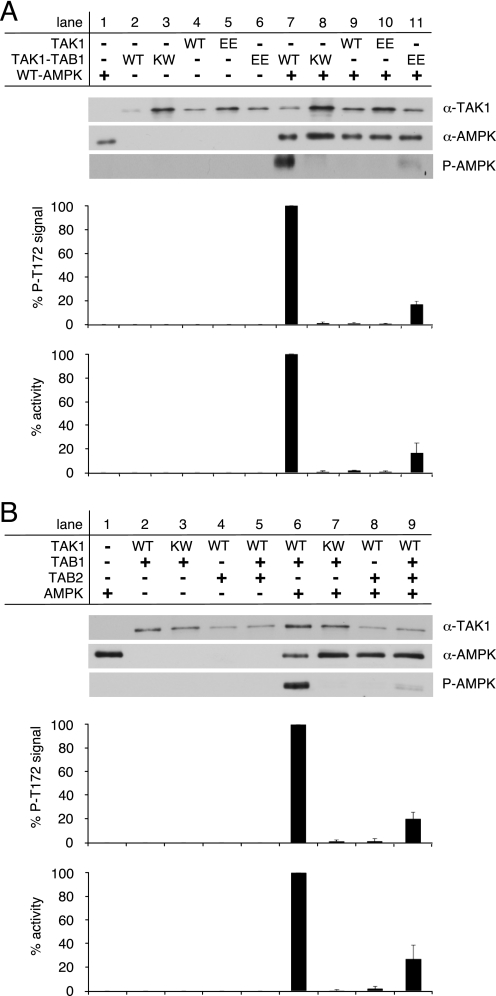
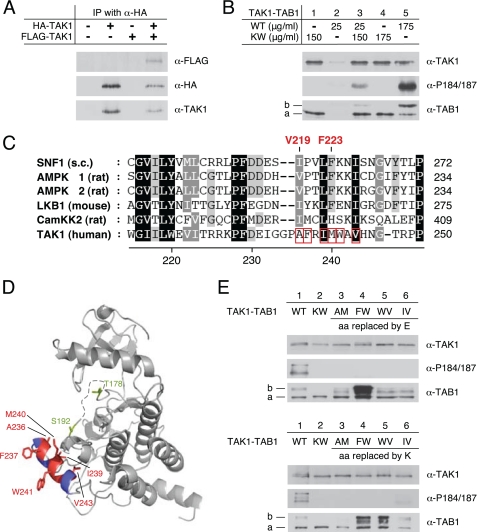
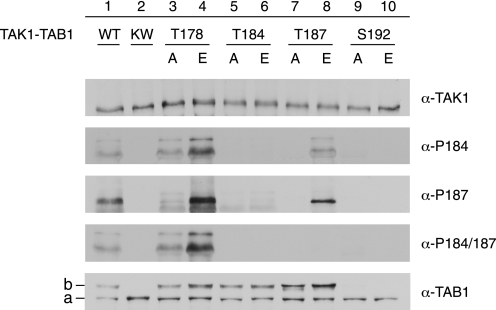
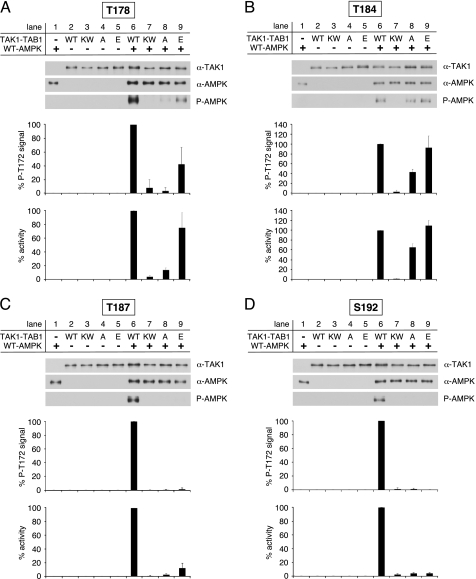
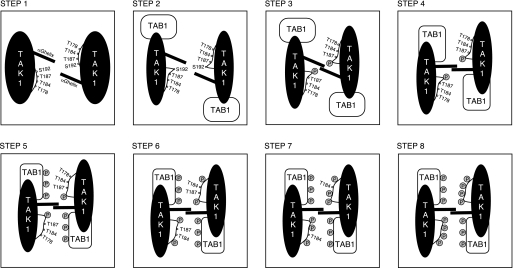
Transforming growth factor-beta-activated kinase 1 (TAK1), an MAP3K, is a key player in processing a multitude of inflammatory stimuli. TAK1 autoactivation involves the interplay with TAK1-binding proteins (TAB), e.g. TAB1 and TAB2, and phosphorylation of several activation segment residues. However, the TAK1 autoactivation is not yet fully understood on the molecular level due to the static nature of available x-ray structural data and the complexity of cellular systems applied for investigation. Here, we established a bacterial expression system to generate recombinant mammalian TAK1 complexes. Co-expression of TAK1 and TAB1, but not TAB2, resulted in a functional and active TAK1-TAB1 complex capable of directly activating full-length heterotrimeric mammalian AMP-activated protein kinase (AMPK) in vitro. TAK1-dependent AMPK activation was mediated via hydrophobic residues of the AMPK kinase domain alphaG-helix as observed in vitro and in transfected cell culture. Co-immunoprecipitation of differently epitope-tagged TAK1 from transfected cells and mutation of hydrophobic alphaG-helix residues in TAK1 point to an intermolecular mechanism of TAB1-induced TAK1 autoactivation, as TAK1 autophosphorylation of the activation segment was impaired in these mutants. TAB1 phosphorylation was enhanced in a subset of these mutants, indicating a critical role of alphaG-helix residues in this process. Analyses of phosphorylation site mutants of the activation segment indicate that autophosphorylation of Ser-192 precedes TAB1 phosphorylation and is followed by sequential phosphorylation of Thr-178, Thr-187, and finally Thr-184. Finally, we present a model for the chronological order of events governing TAB1-induced TAK1 autoactivation.
Figures
References
-
- Yamaguchi K., Shirakabe K., Shibuya H., Irie K., Oishi I., Ueno N., Taniguchi T., Nishida E., Matsumoto K. (1995) Science 270, 2008–2011 - PubMed
-
- Ninomiya-Tsuji J., Kishimoto K., Hiyama A., Inoue J., Cao Z., Matsumoto K. (1999) Nature 398, 252–256 - PubMed
-
- Wang C., Deng L., Hong M., Akkaraju G. R., Inoue J., Chen Z. J. (2001) Nature 412, 346–351 - PubMed
-
- Takaesu G., Surabhi R. M., Park K. J., Ninomiya-Tsuji J., Matsumoto K., Gaynor R. B. (2003) J. Mol. Biol. 326, 105–115 - PubMed
-
- Lee J., Mira-Arbibe L., Ulevitch R. J. (2000) J. Leukocyte Biol. 68, 909–915 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
