

Genetic depletion of the malin E3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin
- PMID: 20538597
- PMCID: PMC2919100
- DOI: 10.1074/jbc.M110.148668
Genetic depletion of the malin E3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin
Abstract
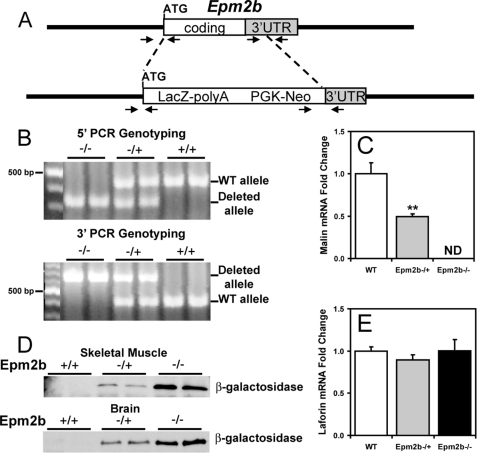
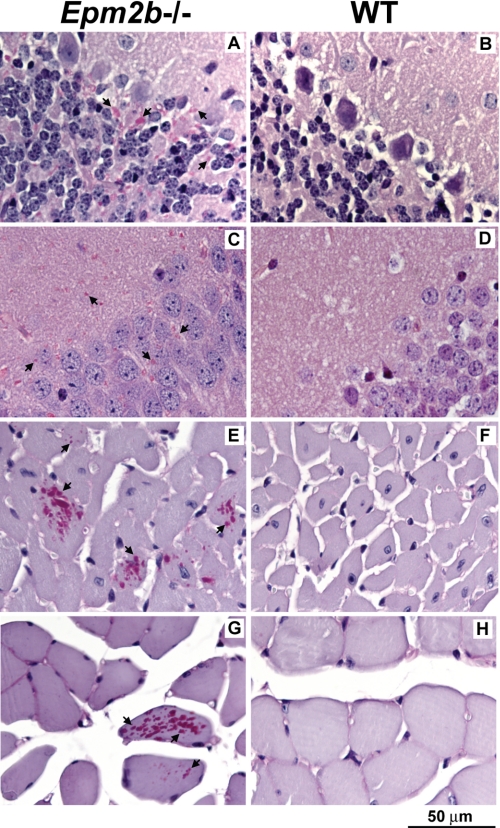
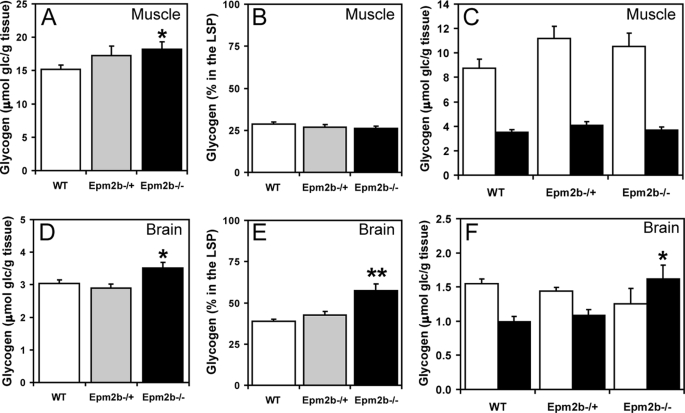
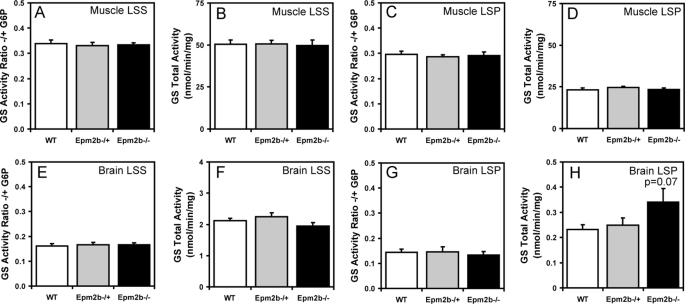
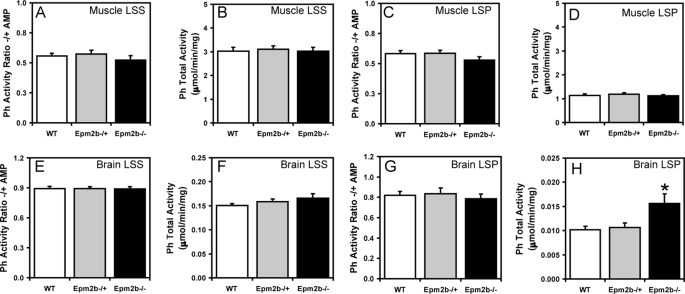
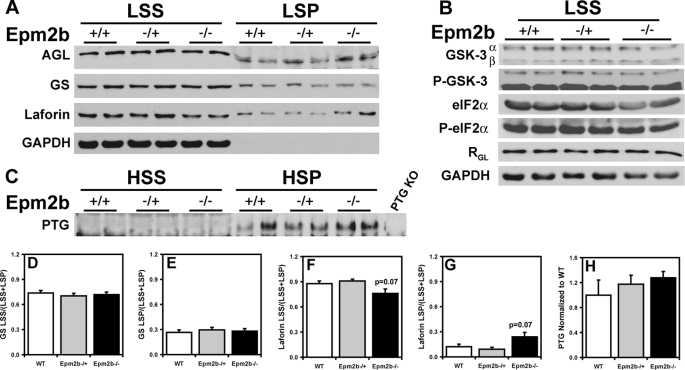
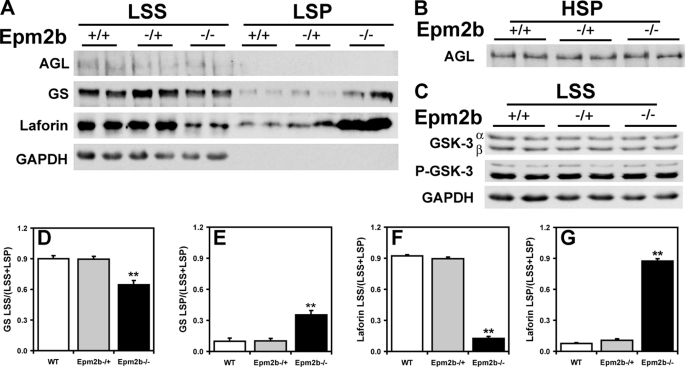
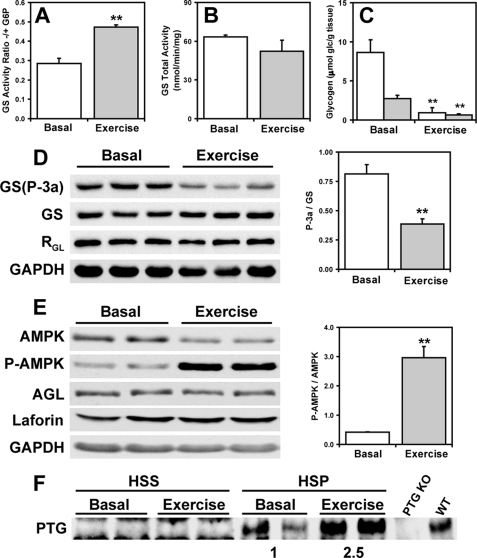
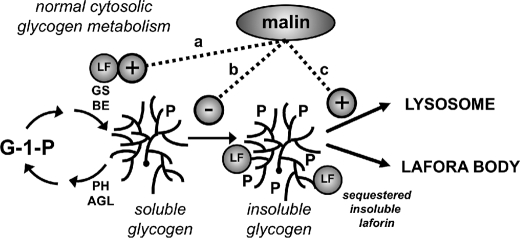
Approximately 90% of cases of Lafora disease, a fatal teenage-onset progressive myoclonus epilepsy, are caused by mutations in either the EPM2A or the EPM2B genes that encode, respectively, a glycogen phosphatase called laforin and an E3 ubiquitin ligase called malin. Lafora disease is characterized by the formation of Lafora bodies, insoluble deposits containing poorly branched glycogen or polyglucosan, in many tissues including skeletal muscle, liver, and brain. Disruption of the Epm2b gene in mice resulted in viable animals that, by 3 months of age, accumulated Lafora bodies in the brain and to a lesser extent in heart and skeletal muscle. Analysis of muscle and brain of the Epm2b(-/-) mice by Western blotting indicated no effect on the levels of glycogen synthase, PTG (type 1 phosphatase-targeting subunit), or debranching enzyme, making it unlikely that these proteins are targeted for destruction by malin, as has been proposed. Total laforin protein was increased in the brain of Epm2b(-/-) mice and, most notably, was redistributed from the soluble, low speed supernatant to the insoluble low speed pellet, which now contained 90% of the total laforin. This result correlated with elevated insolubility of glycogen and glycogen synthase. Because up-regulation of laforin cannot explain Lafora body formation, we conclude that malin functions to maintain laforin associated with soluble glycogen and that its absence causes sequestration of laforin to an insoluble polysaccharide fraction where it is functionally inert.
Figures
References
-
- Delgado-Escueta A. V. (2007) Curr. Neurol. Neurosci. Rep. 7, 428–433 - PubMed
-
- Ganesh S., Puri R., Singh S., Mittal S., Dubey D. (2006) J. Hum. Genet. 51, 1–8 - PubMed
-
- Ramachandran N., Girard J. M., Turnbull J., Minassian B. A. (2009) Epilepsia 50, Suppl. 5, 29–36 - PubMed
-
- Roach P. J. (2002) Curr. Mol. Med. 2, 101–120 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
