

SoRT2: a tool for sorting genomes and reconstructing phylogenetic trees by reversals, generalized transpositions and translocations
- PMID: 20538651
- PMCID: PMC2896082
- DOI: 10.1093/nar/gkq520
SoRT2: a tool for sorting genomes and reconstructing phylogenetic trees by reversals, generalized transpositions and translocations
Abstract
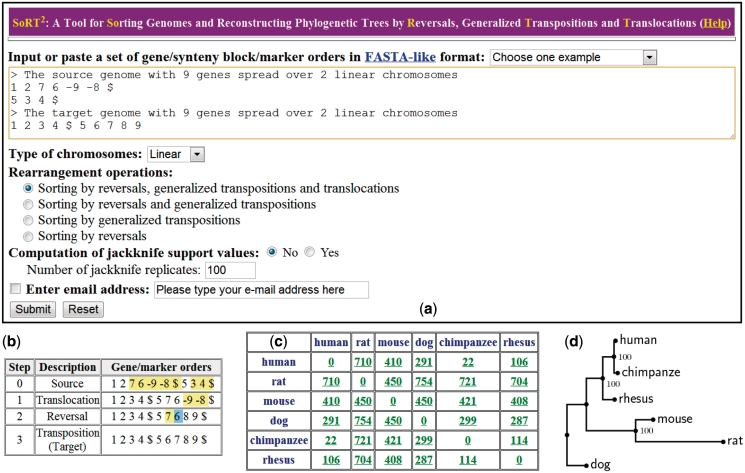
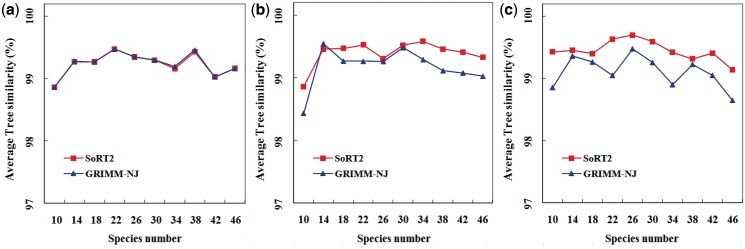
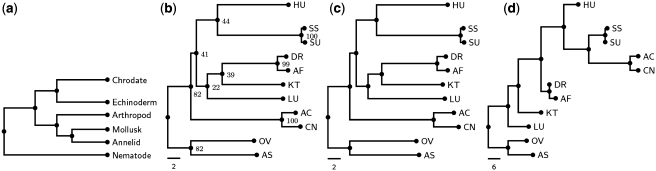
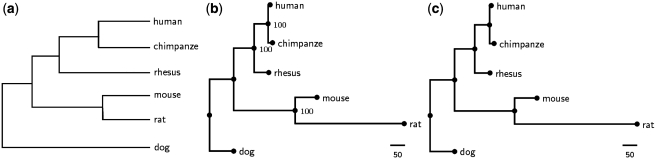
SoRT(2) is a web server that allows the user to perform genome rearrangement analysis involving reversals, generalized transpositions and translocations (including fusions and fissions), and infer phylogenetic trees of genomes being considered based on their pairwise genome rearrangement distances. It takes as input two or more linear/circular multi-chromosomal gene (or synteny block) orders in FASTA-like format. When the input is two genomes, SoRT(2) will quickly calculate their rearrangement distance, as well as a corresponding optimal scenario by highlighting the genes involved in each rearrangement operation. In the case of multiple genomes, SoRT(2) will also construct phylogenetic trees of these genomes based on a matrix of their pairwise rearrangement distances using distance-based approaches, such as neighbor-joining (NJ), unweighted pair group method with arithmetic mean (UPGMA) and Fitch-Margoliash (FM) methods. In addition, if the function of computing jackknife support values is selected, SoRT(2) will further perform the jackknife analysis to evaluate statistical reliability of the constructed NJ, UPGMA and FM trees. SoRT(2) is available online at http://bioalgorithm.life.nctu.edu.tw/SORT2/.
Figures

Similar articles
-
SPRING: a tool for the analysis of genome rearrangement using reversals and block-interchanges.Nucleic Acids Res. 2006 Jul 1;34(Web Server issue):W696-9. doi: 10.1093/nar/gkl169. Nucleic Acids Res. 2006. PMID: 16845100 Free PMC article.
-
Reconstructing genome trees of prokaryotes using overlapping genes.BMC Bioinformatics. 2010 Feb 24;11:102. doi: 10.1186/1471-2105-11-102. BMC Bioinformatics. 2010. PMID: 20181237 Free PMC article.
-
BPhyOG: an interactive server for genome-wide inference of bacterial phylogenies based on overlapping genes.BMC Bioinformatics. 2007 Jul 25;8:266. doi: 10.1186/1471-2105-8-266. BMC Bioinformatics. 2007. PMID: 17650344 Free PMC article.
-
Sorting by weighted reversals, transpositions, and inverted transpositions.J Comput Biol. 2007 Jun;14(5):615-36. doi: 10.1089/cmb.2007.R006. J Comput Biol. 2007. PMID: 17683264 Review.
-
Genome trees and the nature of genome evolution.Annu Rev Microbiol. 2005;59:191-209. doi: 10.1146/annurev.micro.59.030804.121233. Annu Rev Microbiol. 2005. PMID: 16153168 Review.
Cited by
-
Sorting permutations by cut-circularize-linearize-and-paste operations.BMC Genomics. 2011 Nov 30;12 Suppl 3(Suppl 3):S26. doi: 10.1186/1471-2164-12-S3-S26. Epub 2011 Nov 30. BMC Genomics. 2011. PMID: 22369173 Free PMC article.
-
Multiple Lines of Evidence from Mitochondrial Genomes Resolve Phylogenetic Relationships of Parasitic Wasps in Braconidae.Genome Biol Evol. 2016 Sep 4;8(9):2651-62. doi: 10.1093/gbe/evw184. Genome Biol Evol. 2016. PMID: 27503293 Free PMC article.
-
Multi-omics Analysis of Gut Microbiota and Metabolites in Rats With Irritable Bowel Syndrome.Front Cell Infect Microbiol. 2019 May 29;9:178. doi: 10.3389/fcimb.2019.00178. eCollection 2019. Front Cell Infect Microbiol. 2019. PMID: 31192167 Free PMC article.
-
A New Algorithm for Identifying Genome Rearrangements in the Mammalian Evolution.Front Genet. 2019 Oct 29;10:1020. doi: 10.3389/fgene.2019.01020. eCollection 2019. Front Genet. 2019. PMID: 31737036 Free PMC article.
-
Metabolic classification of microbial genomes using functional probes.BMC Genomics. 2012 Apr 27;13:157. doi: 10.1186/1471-2164-13-157. BMC Genomics. 2012. PMID: 22537274 Free PMC article.
References
-
- Blanchette M, Kunisawa T, Sankoff D. Gene order breakpoint evidence in animal mitochondrial phylogeny. J. Mol. Evol. 1999;49:193–203. - PubMed
-
- Cosner ME, Jansen RK, Moret BME, Raubeson LA, Wang L, Warnow T, Wyman S. An empirical comparison of phylogenetic methods on chloroplast gene order data in Campanulaceae. In: Sankoff D, Nadeau JH, editors. Comparative Genomics. London: Kluwer Academic Publishers; 2000. pp. 99–121.
-
- Belda E, Moya A, Silva FJ. Genome rearrangement distances and gene order phylogeny in γ-Proteobacteria. Mol. Biol. Evol. 2005;22:1456–1467. - PubMed
-
- Fertin G, Labarre A, Rusu I, Tannier E, Vialette S. Combinatorics of Genome Rearrangements. Cambridge: The MIT Press; 2009.
