

Hit-and-run stimulation: a novel concept to reactivate latent HIV-1 infection without cytokine gene induction
- PMID: 20538859
- PMCID: PMC2919022
- DOI: 10.1128/JVI.00523-10
Hit-and-run stimulation: a novel concept to reactivate latent HIV-1 infection without cytokine gene induction
Abstract
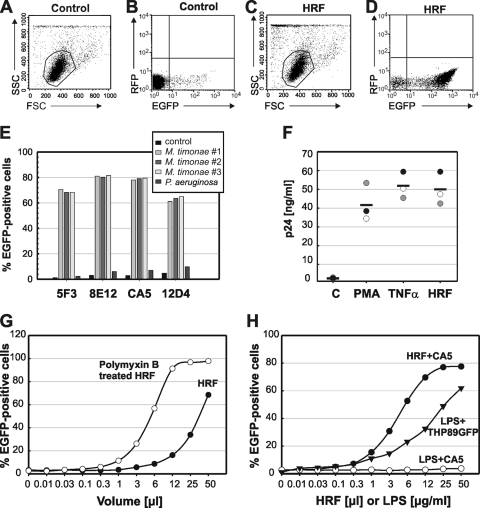
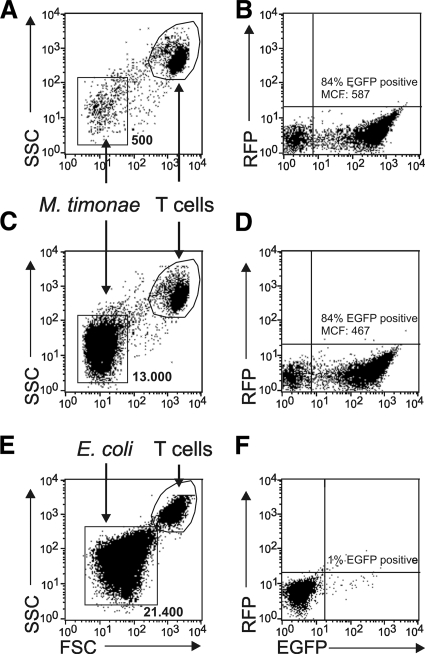
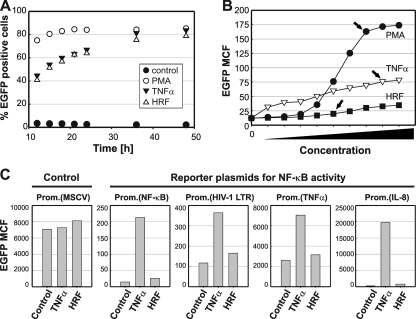
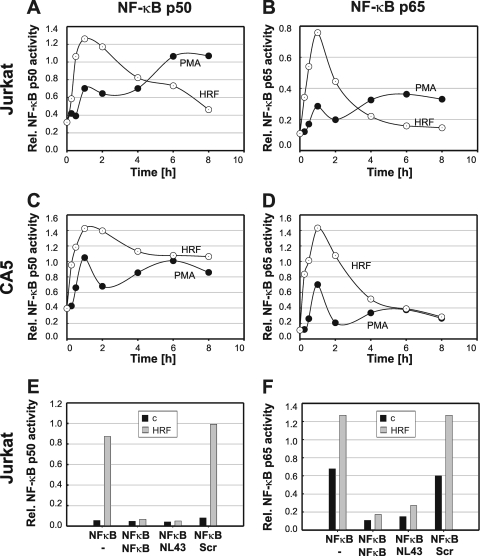
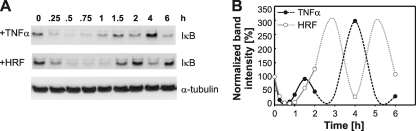
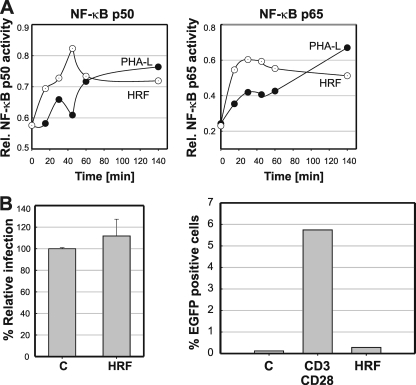
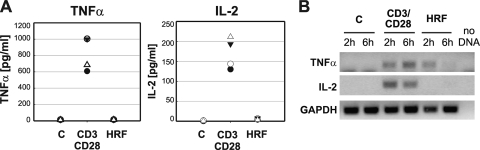
Current antiretroviral therapy (ART) efficiently controls HIV-1 replication but fails to eradicate the virus. Even after years of successful ART, HIV-1 can conceal itself in a latent state in long-lived CD4(+) memory T cells. From this latent reservoir, HIV-1 rebounds during treatment interruptions. Attempts to therapeutically eradicate this viral reservoir have yielded disappointing results. A major problem with previously utilized activating agents is that at the concentrations required for efficient HIV-1 reactivation, these stimuli trigger high-level cytokine gene expression (hypercytokinemia). Therapeutically relevant HIV-1-reactivating agents will have to trigger HIV-1 reactivation without the induction of cytokine expression. We present here a proof-of-principle study showing that this is a possibility. In a high-throughput screening effort, we identified an HIV-1-reactivating protein factor (HRF) secreted by the nonpathogenic bacterium Massilia timonae. In primary T cells and T-cell lines, HRF triggered a high but nonsustained peak of nuclear factor kappa B (NF-kappaB) activity. While this short NF-kappaB peak potently reactivated latent HIV-1 infection, it failed to induce gene expression of several proinflammatory NF-kappaB-dependent cellular genes, such as those for tumor necrosis factor alpha (TNF-alpha), interleukin-8 (IL-8), and gamma interferon (IFN-gamma). Dissociation of cellular and viral gene induction was achievable, as minimum amounts of Tat protein, synthesized following application of a short NF-kappaB pulse, triggered HIV-1 transactivation and subsequent self-perpetuated HIV-1 expression. In the absence of such a positive feedback mechanism, cellular gene expression was not sustained, suggesting that strategies modulating the NF-kappaB activity profile could be used to selectively trigger HIV-1 reactivation.
Figures
Similar articles
-
Alternate NF-κB-Independent Signaling Reactivation of Latent HIV-1 Provirus.J Virol. 2019 Aug 28;93(18):e00495-19. doi: 10.1128/JVI.00495-19. Print 2019 Sep 15. J Virol. 2019. PMID: 31243131 Free PMC article.
-
Maraviroc Is Associated with Latent HIV-1 Reactivation through NF-κB Activation in Resting CD4+ T Cells from HIV-Infected Individuals on Suppressive Antiretroviral Therapy.J Virol. 2018 Apr 13;92(9):e01931-17. doi: 10.1128/JVI.01931-17. Print 2018 May 1. J Virol. 2018. PMID: 29444937 Free PMC article.
-
Posttranscriptional Regulation of HIV-1 Gene Expression during Replication and Reactivation from Latency by Nuclear Matrix Protein MATR3.mBio. 2018 Nov 13;9(6):e02158-18. doi: 10.1128/mBio.02158-18. mBio. 2018. PMID: 30425153 Free PMC article.
-
Efficient Non-Epigenetic Activation of HIV Latency through the T-Cell Receptor Signalosome.Viruses. 2020 Aug 8;12(8):868. doi: 10.3390/v12080868. Viruses. 2020. PMID: 32784426 Free PMC article. Review.
-
A broad drug arsenal to attack a strenuous latent HIV reservoir.Curr Opin Virol. 2019 Oct;38:37-53. doi: 10.1016/j.coviro.2019.06.001. Epub 2019 Jul 16. Curr Opin Virol. 2019. PMID: 31323521 Review.
Cited by
-
Control of HIV latency by epigenetic and non-epigenetic mechanisms.Curr HIV Res. 2011 Dec 1;9(8):554-67. doi: 10.2174/157016211798998736. Curr HIV Res. 2011. PMID: 22211660 Free PMC article.
-
HIV reservoirs and strategies for eradication.Curr HIV/AIDS Rep. 2012 Mar;9(1):5-15. doi: 10.1007/s11904-011-0108-2. Curr HIV/AIDS Rep. 2012. PMID: 22249405 Review.
-
Selected drugs with reported secondary cell-differentiating capacity prime latent HIV-1 infection for reactivation.J Virol. 2012 Sep;86(17):9055-69. doi: 10.1128/JVI.00793-12. Epub 2012 Jun 13. J Virol. 2012. PMID: 22696646 Free PMC article.
-
Kinase control prevents HIV-1 reactivation in spite of high levels of induced NF-κB activity.J Virol. 2012 Apr;86(8):4548-58. doi: 10.1128/JVI.06726-11. Epub 2012 Feb 15. J Virol. 2012. PMID: 22345467 Free PMC article.
-
Promising Role of Toll-Like Receptor 8 Agonist in Concert with Prostratin for Activation of Silent HIV.J Virol. 2017 Jan 31;91(4):e02084-16. doi: 10.1128/JVI.02084-16. Print 2017 Feb 15. J Virol. 2017. PMID: 27928016 Free PMC article.
References
-
- Chun, T. W., L. Carruth, D. Finzi, X. Shen, J. A. DiGiuseppe, H. Taylor, M. Hermankova, K. Chadwick, J. Margolick, T. C. Quinn, Y. H. Kuo, R. Brookmeyer, M. A. Zeiger, P. Barditch-Crovo, and R. F. Siliciano. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183-188. - PubMed
-
- Chun, T. W., R. T. Davey, Jr., M. Ostrowski, J. S. Justement, D. Engel, J. I. Mullins, and A. S. Fauci. 2000. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat. Med. 6:757-761. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
