

Mutant HSPB8 causes motor neuron-specific neurite degeneration
- PMID: 20538880
- PMCID: PMC2908473
- DOI: 10.1093/hmg/ddq234
Mutant HSPB8 causes motor neuron-specific neurite degeneration
Abstract
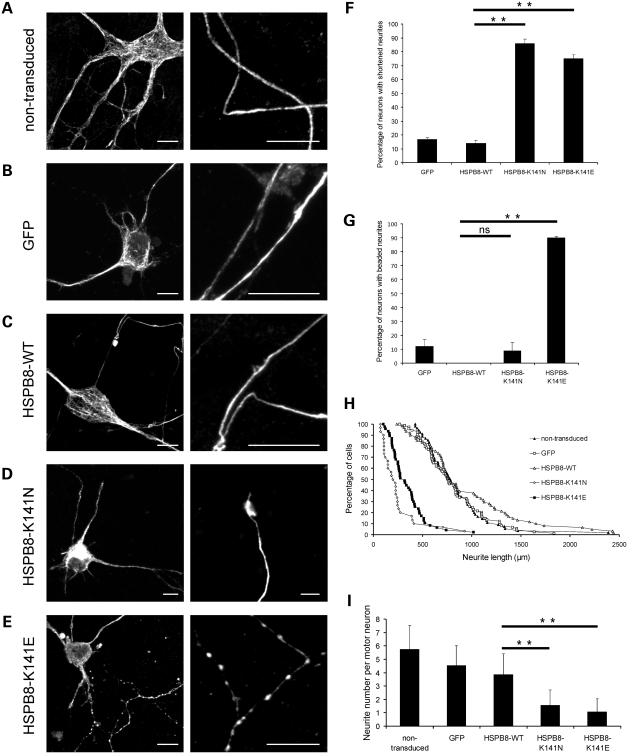
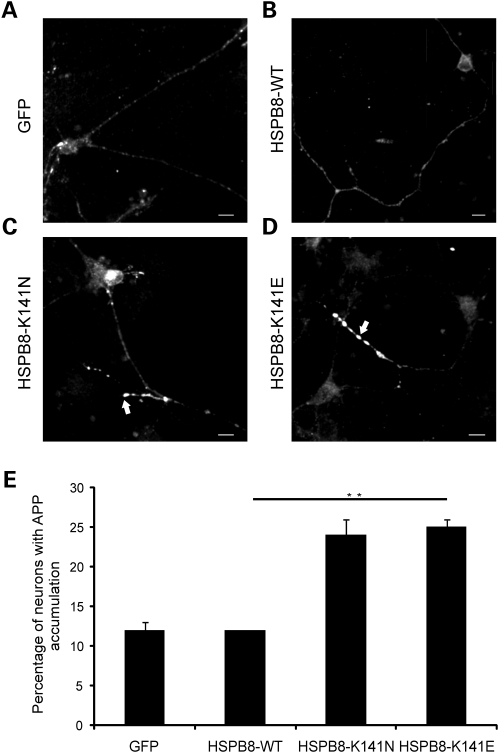
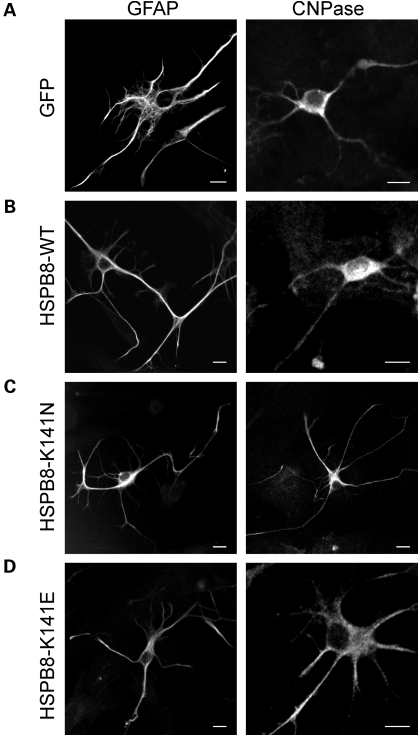
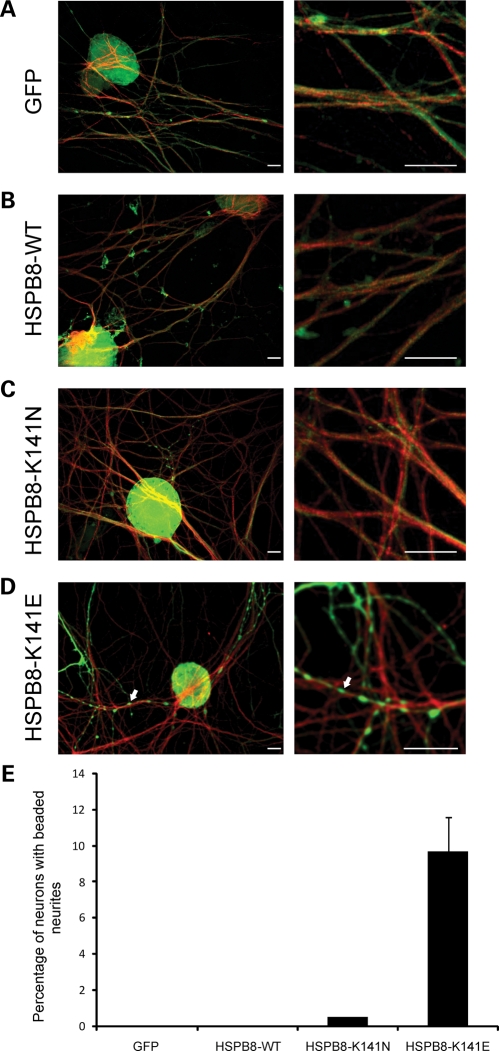
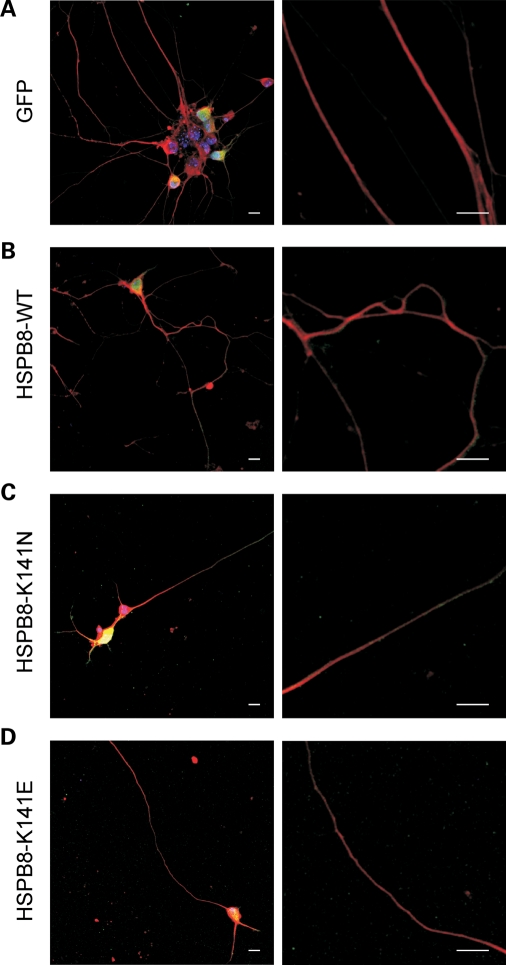
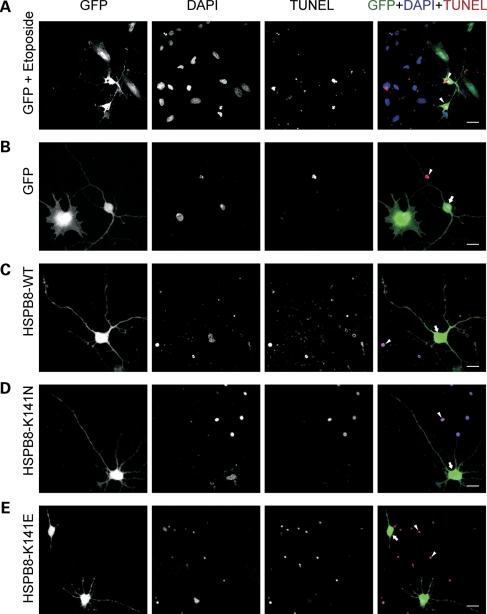
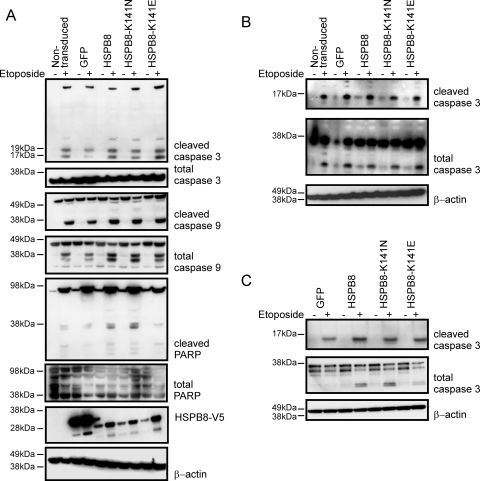
Missense mutations (K141N and K141E) in the alpha-crystallin domain of the small heat shock protein HSPB8 (HSP22) cause distal hereditary motor neuropathy (distal HMN) or Charcot-Marie-Tooth neuropathy type 2L (CMT2L). The mechanism through which mutant HSPB8 leads to a specific motor neuron disease phenotype is currently unknown. To address this question, we compared the effect of mutant HSPB8 in primary neuronal and glial cell cultures. In motor neurons, expression of both HSPB8 K141N and K141E mutations clearly resulted in neurite degeneration, as manifested by a reduction in number of neurites per cell, as well as in a reduction in average length of the neurites. Furthermore, expression of the K141E (and to a lesser extent, K141N) mutation also induced spheroids in the neurites. We did not detect any signs of apoptosis in motor neurons, showing that mutant HSPB8 resulted in neurite degeneration without inducing neuronal death. While overt in motor neurons, these phenotypes were only very mildly present in sensory neurons and completely absent in cortical neurons. Also glial cells did not show an altered phenotype upon expression of mutant HSPB8. These findings show that despite the ubiquitous presence of HSPB8, only motor neurons appear to be affected by the K141N and K141E mutations which explain the predominant motor neuron phenotype in distal HMN and CMT2L.
Figures
References
-
- Dierick I., Irobi J., De Jonghe P., Timmerman V. Small heat shock proteins in inherited peripheral neuropathies. Ann. Med. 2005;37:413–422. doi:10.1080/07853890500296410. - DOI - PubMed
-
- Haslbeck M., Franzmann T., Weinfurtner D., Buchner J. Some like it hot: the structure and function of small heat-shock proteins. Nat. Struct. Mol. Biol. 2005;12:842–846. doi:10.1038/nsmb993. - DOI - PubMed
-
- Walter S., Buchner J. Molecular chaperones—cellular machines for protein folding. Angew. Chem. Int. Ed Engl. 2002;41:1098–1113. doi:10.1002/1521-3773(20020402)41:7<1098::AID-ANIE1098>3.0.CO;2-9. - DOI - PubMed
-
- Tang B.S., Zhao G.H., Luo W., Xia K., Cai F., Pan Q., Zhang R.X., Zhang F.F., Liu X.M., Chen B., et al. Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2L. Hum. Genet. 2005;116:222–224. doi:10.1007/s00439-004-1218-3. - DOI - PubMed
-
- Irobi J., Van Impe K., Seeman P., Jordanova A., Dierick I., Verpoorten N., Michalik A., De Vriendt E., Jacobs A., Van Gerwen V., et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat. Genet. 2004;36:597–601. doi:10.1038/ng1328. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
