

When is chemical similarity significant? The statistical distribution of chemical similarity scores and its extreme values
- PMID: 20540577
- PMCID: PMC2914517
- DOI: 10.1021/ci100010v
When is chemical similarity significant? The statistical distribution of chemical similarity scores and its extreme values
Abstract
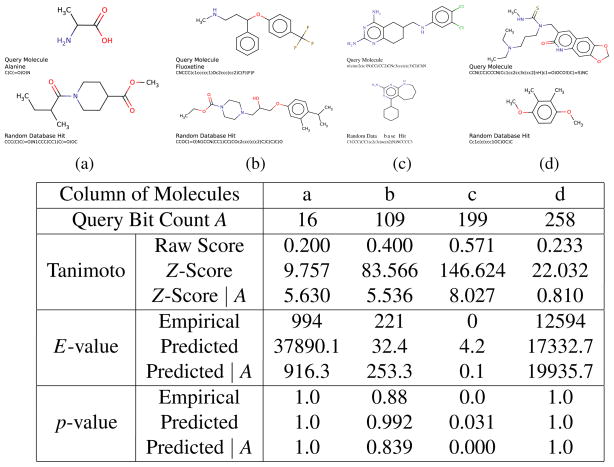
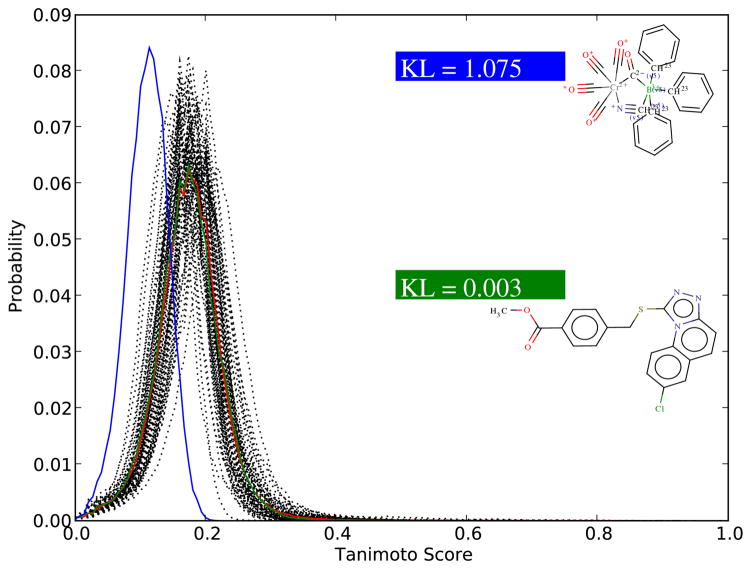
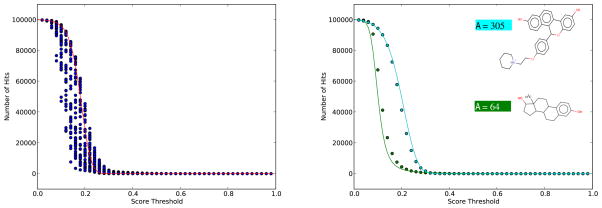
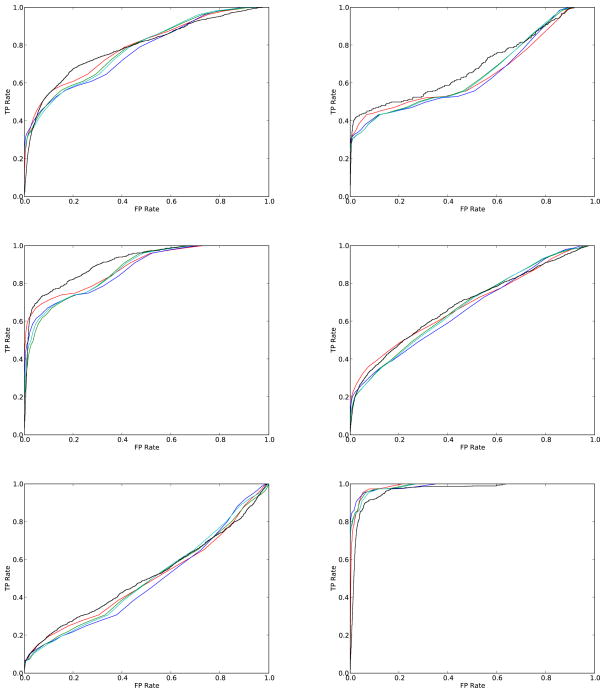
As repositories of chemical molecules continue to expand and become more open, it becomes increasingly important to develop tools to search them efficiently and assess the statistical significance of chemical similarity scores. Here, we develop a general framework for understanding, modeling, predicting, and approximating the distribution of chemical similarity scores and its extreme values in large databases. The framework can be applied to different chemical representations and similarity measures but is demonstrated here using the most common binary fingerprints with the Tanimoto similarity measure. After introducing several probabilistic models of fingerprints, including the Conditional Gaussian Uniform model, we show that the distribution of Tanimoto scores can be approximated by the distribution of the ratio of two correlated Normal random variables associated with the corresponding unions and intersections. This remains true also when the distribution of similarity scores is conditioned on the size of the query molecules to derive more fine-grained results and improve chemical retrieval. The corresponding extreme value distributions for the maximum scores are approximated by Weibull distributions. From these various distributions and their analytical forms, Z-scores, E-values, and p-values are derived to assess the significance of similarity scores. In addition, the framework also allows one to predict the value of standard chemical retrieval metrics, such as sensitivity and specificity at fixed thresholds, or receiver operating characteristic (ROC) curves at multiple thresholds, and to detect outliers in the form of atypical molecules. Numerous and diverse experiments that have been performed, in part with large sets of molecules from the ChemDB, show remarkable agreement between theory and empirical results.
Figures
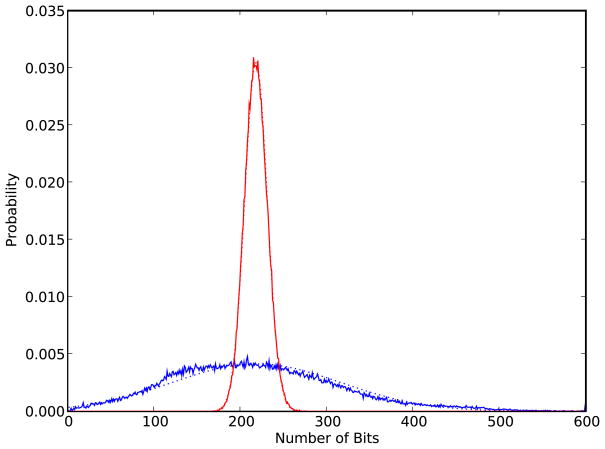
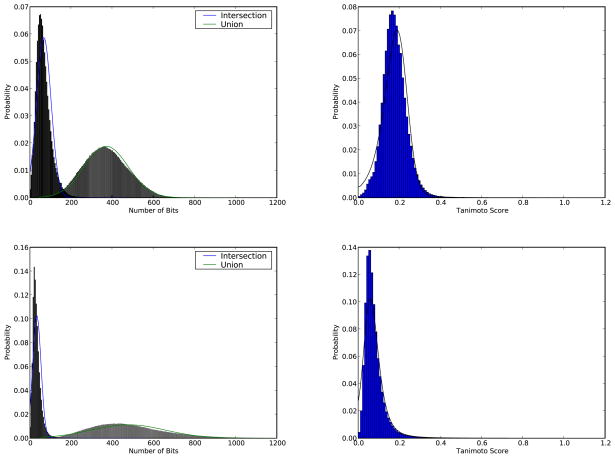
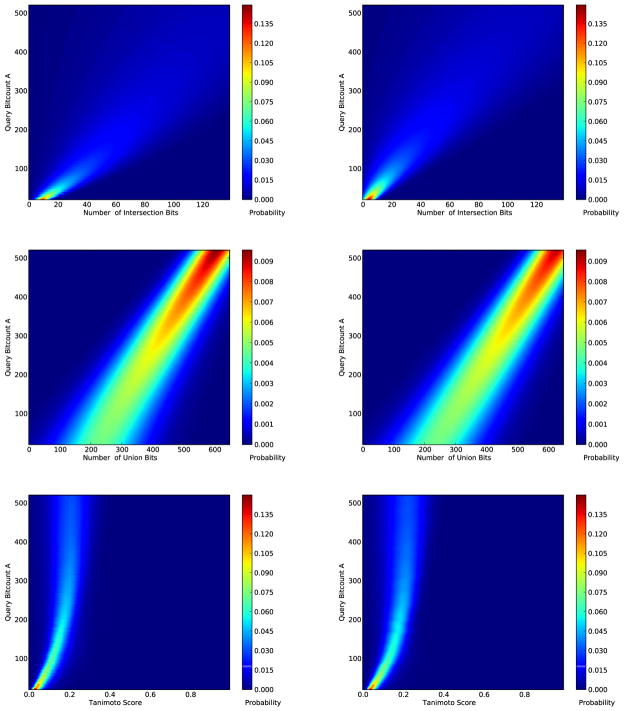
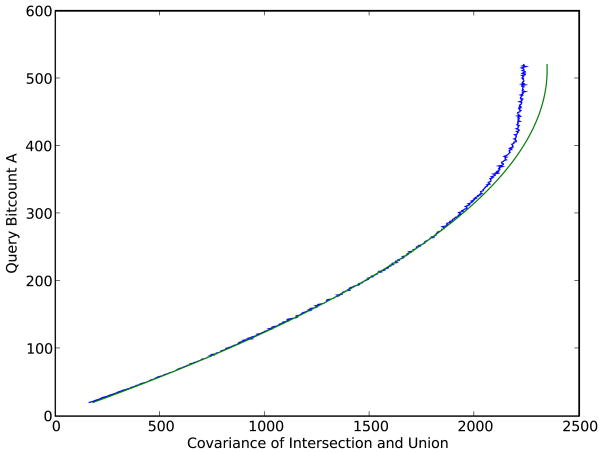
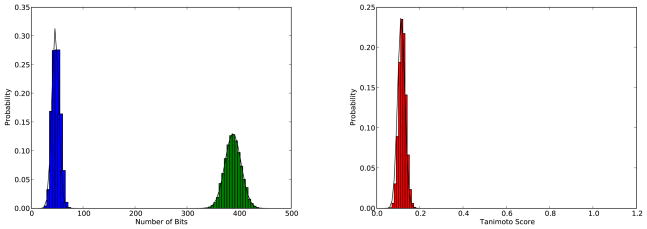
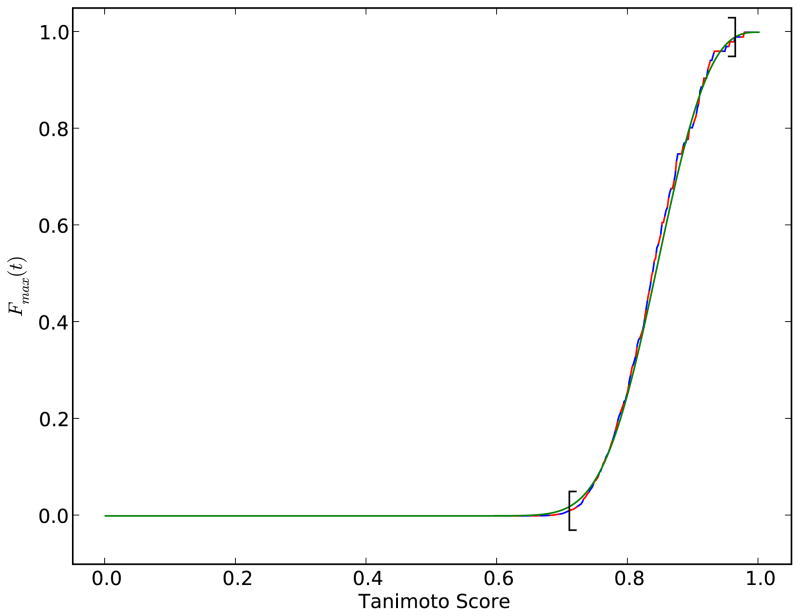
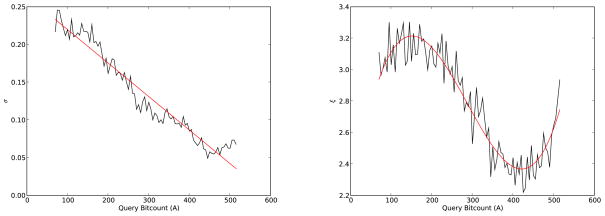
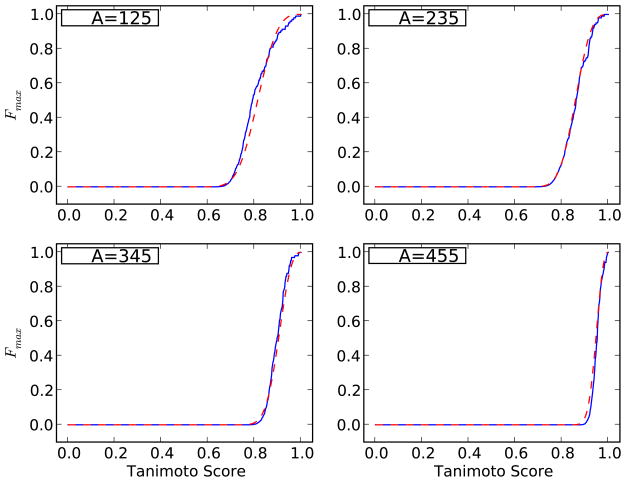
References
-
- Chen J, Swamidass SJ, Dou Y, Bruand J, Baldi P. ChemDB: a public database of small molecules and related chemoinformatics resources. Bioinformatics. 2005;21:4133–4139. - PubMed
-
- Chen J, Linstead E, Swamidass SJ, Wang D, Baldi P. ChemDB Update–Full Text Search and Virtual Chemical Space. Bioinformatics. 2007;23:2348–2351. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
