

Identification of BERP (brain-expressed RING finger protein) as a p53 target gene that modulates seizure susceptibility through interacting with GABA(A) receptors
- PMID: 20543135
- PMCID: PMC2900672
- DOI: 10.1073/pnas.1006529107
Identification of BERP (brain-expressed RING finger protein) as a p53 target gene that modulates seizure susceptibility through interacting with GABA(A) receptors
Abstract
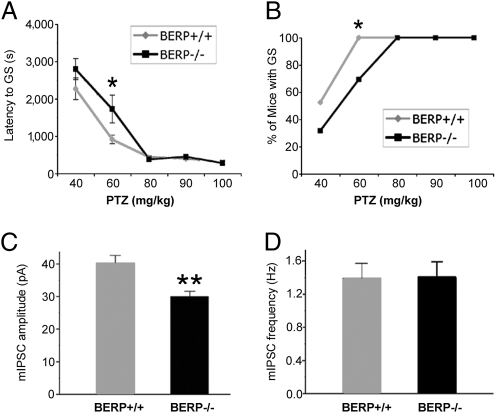
p53 is a central player in responses to cellular stresses and a major tumor suppressor. The identification of unique molecules within the p53 signaling network can reveal functions of this important transcription factor. Here, we show that brain-expressed RING finger protein (BERP) is a gene whose expression is up-regulated in a p53-dependent manner in human cells and in mice. We generated BERP-deficient mice by gene targeting and demonstrated that they exhibit increased resistance to pentylenetetrazol-induced seizures. Electrophysiological and biochemical studies of cultured cortical neurons of BERP-deficient mice showed a decrease in the amplitude of GABA(A) receptor (GABA(A)R)-mediated miniature inhibitory postsynaptic currents as well as reduced surface protein expression of GABA(A)Rs containing the gamma2-subunit. However, BERP deficiency did not decrease GABA(A)Rgamma2 mRNA levels, raising the possibility that BERP may act at a posttranscriptional level to regulate the intracellular trafficking of GABA(A)Rs. Our results indicate that BERP is a unique p53-regulated gene and suggest a role for p53 within the central nervous system.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




Similar articles
-
Genetic inactivation of the sigma-1 chaperone protein results in decreased expression of the R2 subunit of the GABA-B receptor and increased susceptibility to seizures.Neurobiol Dis. 2021 Mar;150:105244. doi: 10.1016/j.nbd.2020.105244. Epub 2020 Dec 30. Neurobiol Dis. 2021. PMID: 33385516
-
[Long-term effects of neonatal recurrent seizures on gamma-aminobutyric acid A receptor alpha1 and gamma2 subunit expressions in the rat brain].Beijing Da Xue Xue Bao Yi Xue Ban. 2006 Dec 18;38(6):628-33. Beijing Da Xue Xue Bao Yi Xue Ban. 2006. PMID: 17173085 Chinese.
-
Egr3 stimulation of GABRA4 promoter activity as a mechanism for seizure-induced up-regulation of GABA(A) receptor alpha4 subunit expression.Proc Natl Acad Sci U S A. 2005 Aug 16;102(33):11894-9. doi: 10.1073/pnas.0501434102. Epub 2005 Aug 9. Proc Natl Acad Sci U S A. 2005. PMID: 16091474 Free PMC article.
-
Overexpressing wild-type γ2 subunits rescued the seizure phenotype in Gabrg2+/Q390X Dravet syndrome mice.Epilepsia. 2017 Aug;58(8):1451-1461. doi: 10.1111/epi.13810. Epub 2017 Jun 6. Epilepsia. 2017. PMID: 28586508 Free PMC article.
-
Cloning and characterization of a gene (RNF22) encoding a novel brain expressed ring finger protein (BERP) that maps to human chromosome 11p15.5.Genomics. 2001 Feb 1;71(3):363-7. doi: 10.1006/geno.2000.6452. Genomics. 2001. PMID: 11170753
Cited by
-
GABAA receptor trafficking-mediated plasticity of inhibitory synapses.Neuron. 2011 May 12;70(3):385-409. doi: 10.1016/j.neuron.2011.03.024. Neuron. 2011. PMID: 21555068 Free PMC article. Review.
-
It's a TRIM-endous view from the top: the varied roles of TRIpartite Motif proteins in brain development and disease.Front Mol Neurosci. 2023 Dec 5;16:1287257. doi: 10.3389/fnmol.2023.1287257. eCollection 2023. Front Mol Neurosci. 2023. PMID: 38115822 Free PMC article. Review.
-
Dual roles of TRIM3 in colorectal cancer by retaining p53 in the cytoplasm to decrease its nuclear expression.Cell Death Discov. 2023 Mar 9;9(1):85. doi: 10.1038/s41420-023-01386-1. Cell Death Discov. 2023. PMID: 36894560 Free PMC article.
-
The ubiquitin ligase LIN41/TRIM71 targets p53 to antagonize cell death and differentiation pathways during stem cell differentiation.Cell Death Differ. 2017 Jun;24(6):1063-1078. doi: 10.1038/cdd.2017.54. Epub 2017 Apr 21. Cell Death Differ. 2017. PMID: 28430184 Free PMC article.
-
Metabolic regulation by p53.J Mol Med (Berl). 2011 Mar;89(3):237-45. doi: 10.1007/s00109-011-0735-5. Epub 2011 Feb 23. J Mol Med (Berl). 2011. PMID: 21340684 Free PMC article. Review.
References
-
- Vousden KH, Lu X. Live or let die: The cell's response to p53. Nat Rev Cancer. 2002;2:594–604. - PubMed
-
- Levine AJ, Hu W, Feng Z. The P53 pathway: What questions remain to be explored? Cell Death Differ. 2006;13:1027–1036. - PubMed
-
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. - PubMed
-
- Daujat S, Neel H, Piette J. MDM2: Life without p53. Trends Genet. 2001;17:459–464. - PubMed
-
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
