

Oncolytic Viruses for Cancer Therapy: Overcoming the Obstacles
- PMID: 20543907
- PMCID: PMC2883714
- DOI: 10.3390/v2010078
Oncolytic Viruses for Cancer Therapy: Overcoming the Obstacles
Abstract
Targeted therapy of cancer using oncolytic viruses has generated much interest over the past few years in the light of the limited efficacy and side effects of standard cancer therapeutics for advanced disease. In 2006, the world witnessed the first government-approved oncolytic virus for the treatment of head and neck cancer. It has been known for many years that viruses have the ability to replicate in and lyse cancer cells. Although encouraging results have been demonstrated in vitro and in animal models, most oncolytic viruses have failed to impress in the clinical setting. The explanation is multifactorial, determined by the complex interactions between the tumor and its microenvironment, the virus, and the host immune response. This review focuses on discussion of the obstacles that oncolytic virotherapy faces and recent advances made to overcome them, with particular reference to adenoviruses.
Figures
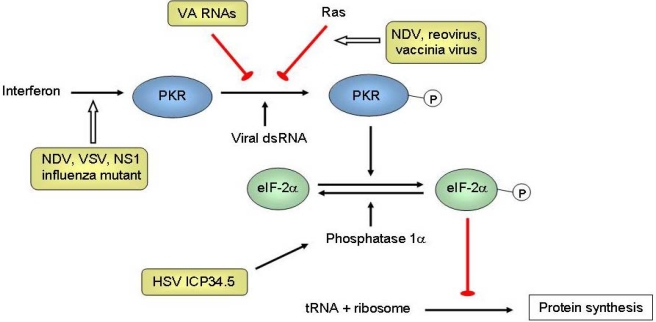
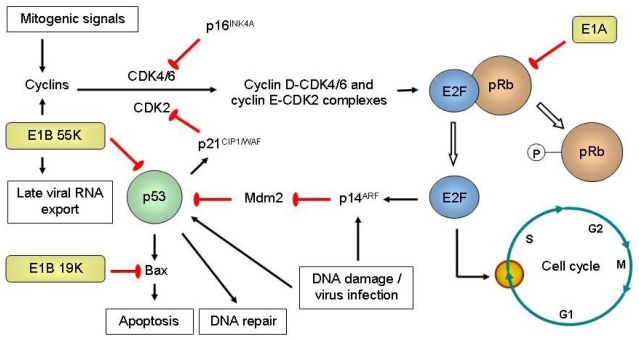
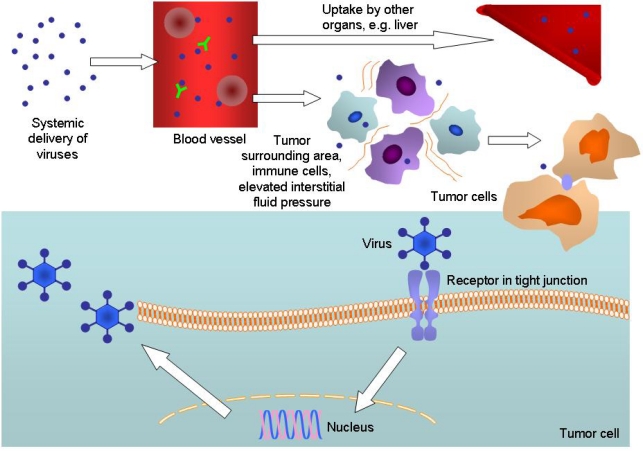
References
-
- Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15:651–659. - PubMed
-
- Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252:854–856. - PubMed
-
- Garber K. China approves world's first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst. 2006;98:298–300. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
