

Novel diagnostic features of dysferlinopathies
- PMID: 20544924
- PMCID: PMC3025537
- DOI: 10.1002/mus.21650
Novel diagnostic features of dysferlinopathies
Abstract
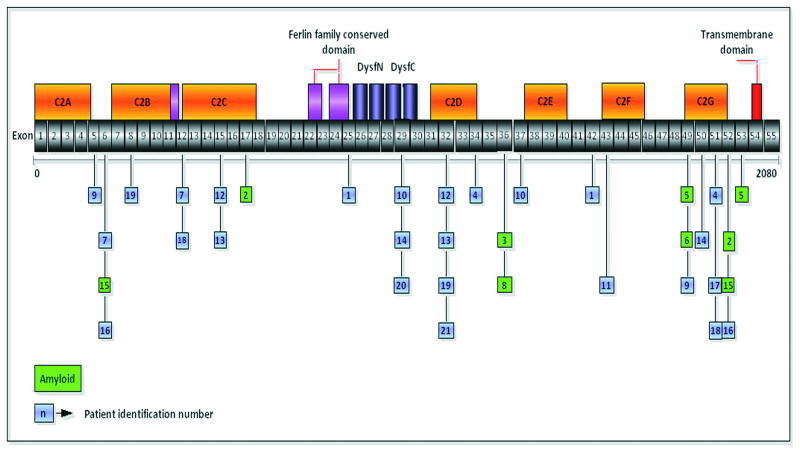
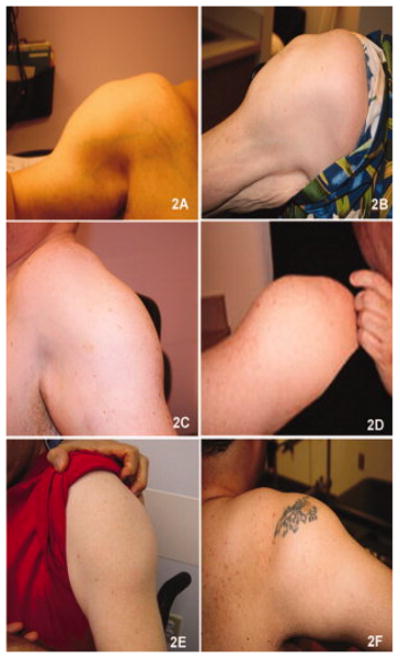
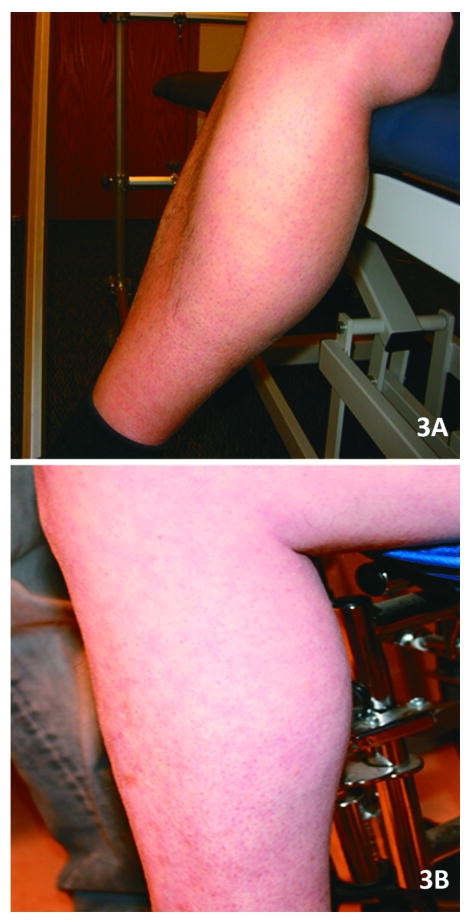
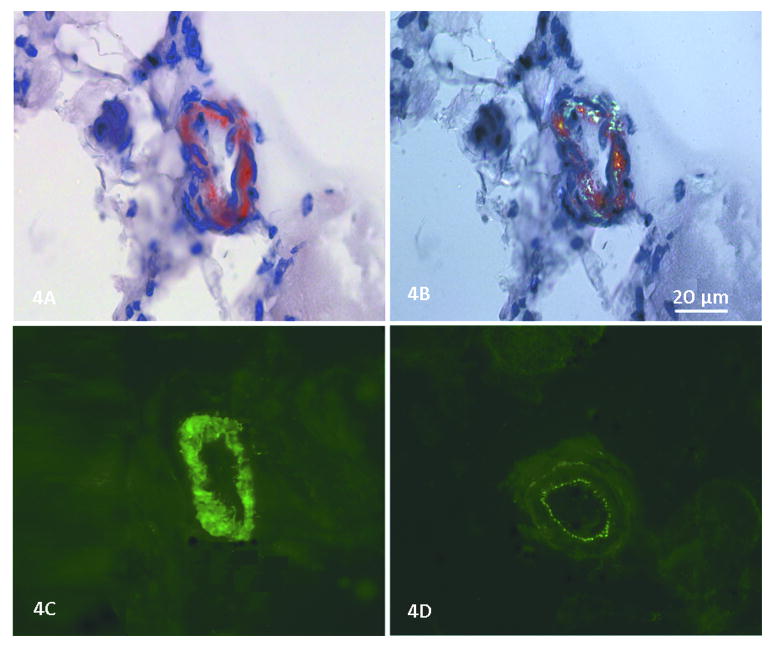
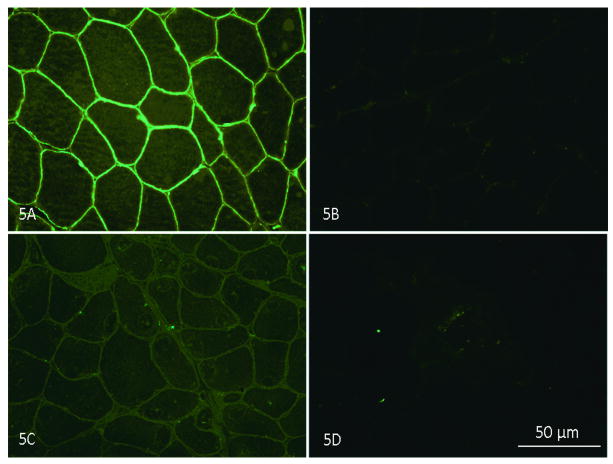
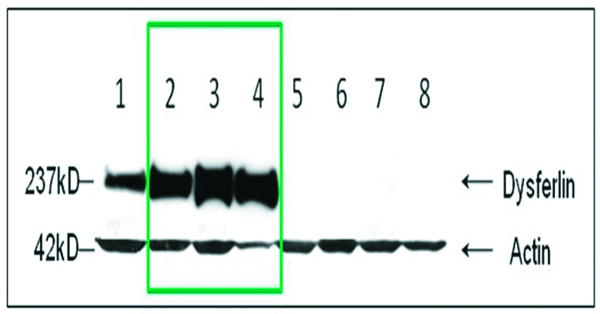
Reports of dysferlinopathy have suggested a clinically heterogeneous group of patients. We identified specific novel molecular and phenotypic features that help distinguish dysferlinopathies from other forms of limb-girdle muscular dystrophy (LGMD). A detailed history, physical exam, and protein and mutation analysis of genomic DNA was done for all subjects. Five of 21 confirmed DYSF gene mutations were not previously reported. A distinct "bulge" of the deltoid muscle in combination with other findings was a striking feature in all patients. Six subjects had atypical calf enlargement, and 3 of these exhibited a paradoxical pattern of dysferlin expression: severely reduced by direct immunofluorescence with overexpression on Western blots. Six patients showed amyloid deposits in muscle that extended these findings to new domains of the dysferlin gene, including the C2G domain. Correlative studies showed colocalization of amyloid with deposition of dysferlin. The present data further serve to guide clinicians facing the expensive task of molecular characterization of patients with an LGMD phenotype.
Figures
Similar articles
-
DYSF mutation analysis in a group of Chinese patients with dysferlinopathy.Clin Neurol Neurosurg. 2013 Aug;115(8):1234-7. doi: 10.1016/j.clineuro.2012.11.010. Epub 2012 Dec 14. Clin Neurol Neurosurg. 2013. PMID: 23254335
-
Painful enlargement of the calf muscles in limb girdle muscular dystrophy type 2B (LGMD2B) with a novel compound heterozygous mutation in DYSF.Neuromuscul Disord. 2007 Feb;17(2):157-62. doi: 10.1016/j.nmd.2006.09.015. Epub 2006 Nov 28. Neuromuscul Disord. 2007. PMID: 17129727
-
Chronic pain as a presenting feature of dysferlinopathy.Neuromuscul Disord. 2025 Jan;46:105269. doi: 10.1016/j.nmd.2024.105269. Epub 2024 Dec 14. Neuromuscul Disord. 2025. PMID: 39798170
-
Dysferlin function in skeletal muscle: Possible pathological mechanisms and therapeutical targets in dysferlinopathies.Exp Neurol. 2016 Sep;283(Pt A):246-54. doi: 10.1016/j.expneurol.2016.06.026. Epub 2016 Jun 25. Exp Neurol. 2016. PMID: 27349407 Review.
-
Dysferlinopathies: Clinical and genetic variability.Clin Genet. 2022 Dec;102(6):465-473. doi: 10.1111/cge.14216. Epub 2022 Sep 6. Clin Genet. 2022. PMID: 36029111 Review.
Cited by
-
AAV.Dysferlin Overlap Vectors Restore Function in Dysferlinopathy Animal Models.Ann Clin Transl Neurol. 2015 Mar;2(3):256-70. doi: 10.1002/acn3.172. Epub 2015 Jan 20. Ann Clin Transl Neurol. 2015. PMID: 25815352 Free PMC article.
-
Limb-Girdle Muscular Dystrophies (LGMD): Clinical features, diagnosis and genetic variability through next generation sequencing.Glob Med Genet. 2024 Dec 16;12(1):100035. doi: 10.1016/j.gmg.2024.100035. eCollection 2025 Mar. Glob Med Genet. 2024. PMID: 39925440 Free PMC article.
-
Diagnosis by protein analysis of dysferlinopathy in two patients mistaken as polymyositis.Acta Myol. 2011 Dec;30(3):185-7. Acta Myol. 2011. PMID: 22616201 Free PMC article.
-
The Clinical Outcome Study for dysferlinopathy: An international multicenter study.Neurol Genet. 2016 Aug 4;2(4):e89. doi: 10.1212/NXG.0000000000000089. eCollection 2016 Aug. Neurol Genet. 2016. PMID: 27602406 Free PMC article.
-
The Dysferlinopathies Conundrum: Clinical Spectra, Disease Mechanism and Genetic Approaches for Treatments.Biomolecules. 2024 Feb 21;14(3):256. doi: 10.3390/biom14030256. Biomolecules. 2024. PMID: 38540676 Free PMC article. Review.
References
-
- Guglieri M, Straub V, Bushby K, Lochmuller H. Limb-girdle muscular dystrophies. Curr Opin Neurol. 2008;21:576–584. - PubMed
-
- Guglieri M, Magri F, D'Angelo MG, Prelle A, Morandi L, Rodolico C, et al. Clinical, molecular, and protein correlations in a large sample of genetically diagnosed Italian limb girdle muscular dystrophy patients. Hum Mut. 2008;29:258–266. - PubMed
-
- Jarry J, Rioux MF, Bolduc V, Robitaille Y, Khoury V, Thiffault I, et al. A novel autosomal recessive limb-girdle muscular dystrophy with quadriceps atrophy maps to 11p13-p12. Brain. 2007;130:368–380. - PubMed
-
- Bashir R, Keers S, Strachan T, Passos-Bueno R, Zatz M, Weissenbach J, et al. Genetic and physical mapping at the limb-girdle muscular dystrophy locus (LGMD2B) on chromosome 2p. Genomics. 1996;33:46–52. - PubMed
-
- Bejaoui K, Hirabayashi K, Hentati F, Haines JL, Ben Hamida C, Belal S, et al. Linkage of Miyoshi myopathy (distal autosomal recessive muscular dystrophy) locus to chromosome 2p12-14. Neurology. 1995;45:768–772. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
