

Improving computational protein design by using structure-derived sequence profile
- PMID: 20544969
- PMCID: PMC3058783
- DOI: 10.1002/prot.22746
Improving computational protein design by using structure-derived sequence profile
Abstract
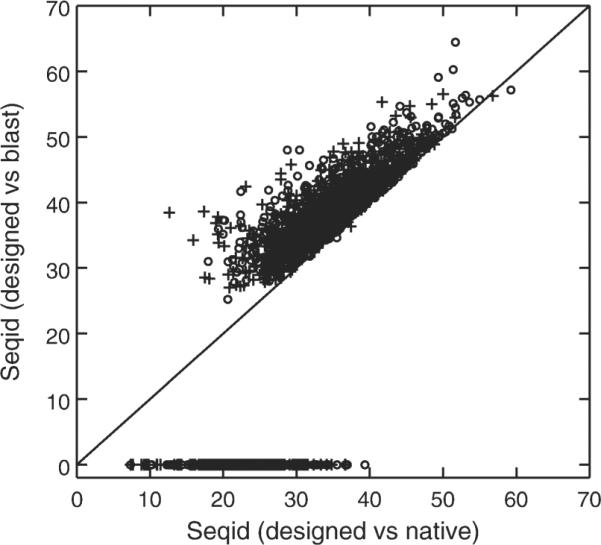
Designing a protein sequence that will fold into a predefined structure is of both practical and fundamental interest. Many successful, computational designs in the last decade resulted from improved understanding of hydrophobic and polar interactions between side chains of amino acid residues in stabilizing protein tertiary structures. However, the coupling between main-chain backbone structure and local sequence has yet to be fully addressed. Here, we attempt to account for such coupling by using a sequence profile derived from the sequences of five residue fragments in a fragment library that are structurally matched to the five-residue segments contained in a target structure. We further introduced a term to reduce low complexity regions of designed sequences. These two terms together with optimized reference states for amino-acid residues were implemented in the RosettaDesign program. The new method, called RosettaDesign-SR, makes a 12% increase (from 34 to 46%) in fraction of proteins whose designed sequences are more than 35% identical to wild-type sequences. Meanwhile, it reduces 8% (from 22% to 14%) to the number of designed sequences that are not homologous to any known protein sequences according to psi-blast. More importantly, the sequences designed by RosettaDesign-SR have 2-3% more polar residues at the surface and core regions of proteins and these surface and core polar residues have about 4% higher sequence identity to wild-type sequences than by RosettaDesign. Thus, the proteins designed by RosettaDesign-SR should be less likely to aggregate and more likely to have unique structures due to more specific polar interactions.
(c) 2010 Wiley-Liss, Inc.
Figures
References
-
- Dahiyat BI, Mayo SL. De novo protein design: fully automated sequence selection. Science. 1997;278:82–87. - PubMed
-
- Harbury PB, Plecs JJ, Tidor B, Alber T, Kim PS. High-resolution protein design with backbone freedom. Science. 1998;282:1462–1467. - PubMed
-
- Shah PS, Hom GK, Ross SA, Lassila JK, Crowhurst KA, Mayo SL. Full-sequence computational design and solution structure of a thermostable protein variant. J Mol Biol. 2007;372:1–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
