

Structural assembly of molecular complexes based on residual dipolar couplings
- PMID: 20550109
- PMCID: PMC2931813
- DOI: 10.1021/ja100447p
Structural assembly of molecular complexes based on residual dipolar couplings
Abstract
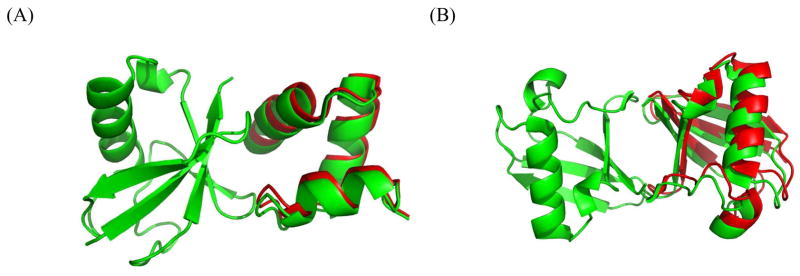
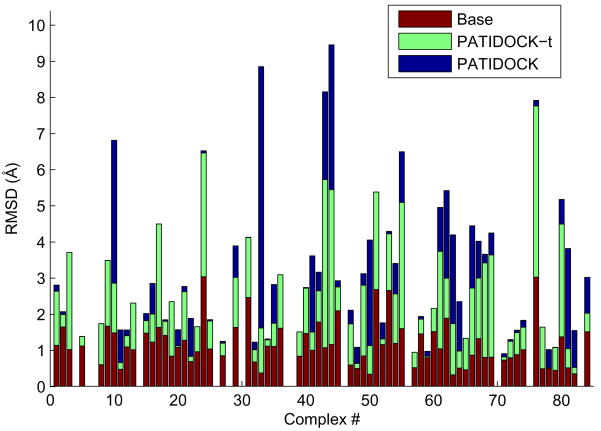
We present and evaluate a rigid-body molecular docking method, called PATIDOCK, that relies solely on the three-dimensional structure of the individual components and the experimentally derived residual dipolar couplings (RDCs) for the complex. We show that, given an accurate ab initio predictor of the alignment tensor from a protein structure, it is possible to accurately assemble a protein-protein complex by utilizing the RDCs' sensitivity to molecular shape to guide the docking. The proposed docking method is robust against experimental errors in the RDCs and computationally efficient. We analyze the accuracy and efficiency of this method using experimental or synthetic RDC data for several proteins, as well as synthetic data for a large variety of protein-protein complexes. We also test our method on two protein systems for which the structure of the complex and steric-alignment data are available (Lys48-linked diubiquitin and a complex of ubiquitin and a ubiquitin-associated domain) and analyze the effect of flexible unstructured tails on the outcome of docking. The results demonstrate that it is fundamentally possible to assemble a protein-protein complex solely on the basis of experimental RDC data and the prediction of the alignment tensor from 3D structures. Thus, despite the purely angular nature of RDCs, they can be converted into intermolecular distance/translational constraints. Additionally, we show a method for combining RDCs with other experimental data, such as ambiguous constraints from interface mapping, to further improve structure characterization of protein complexes.
Figures
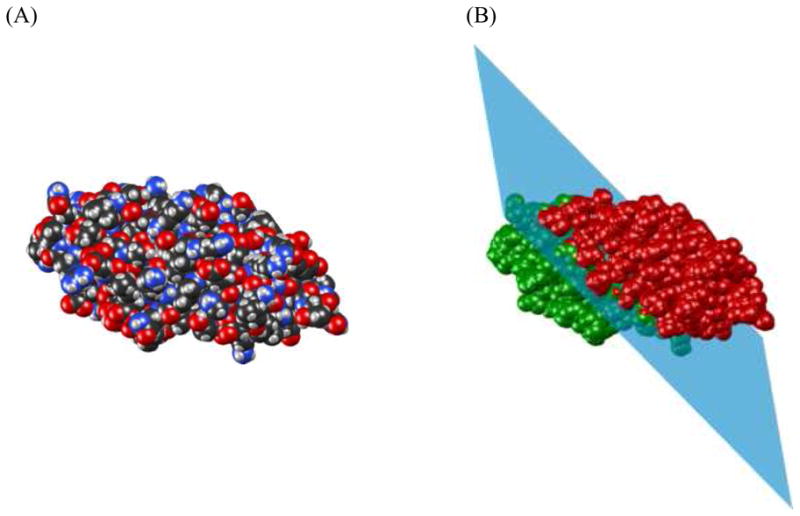
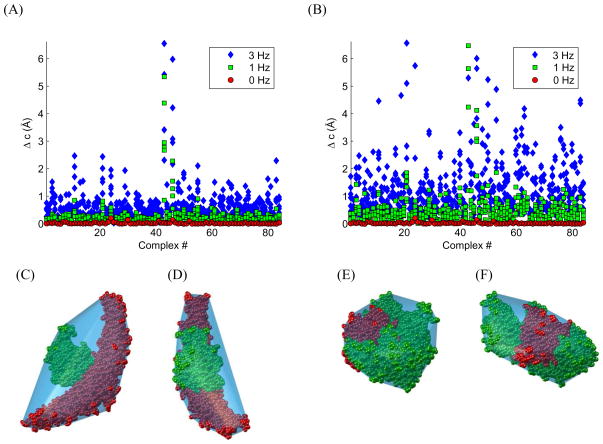
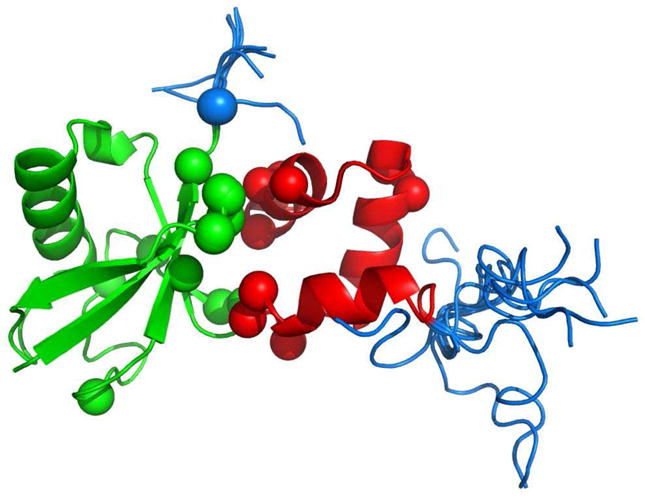

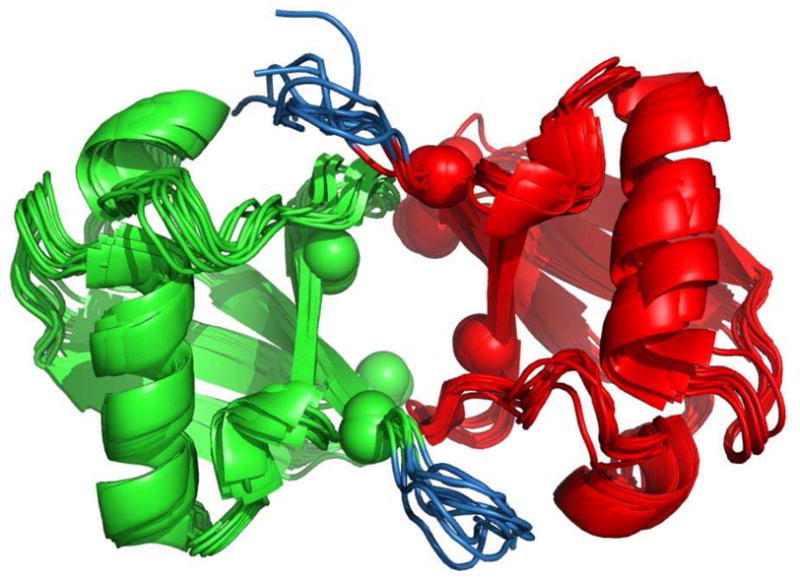
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
