

doi: 10.1186/gb-2010-11-6-r61.
Epub 2010 Jun 15.
Design and evaluation of genome-wide libraries for RNA interference screens
Affiliations
- PMID: 20550664
- PMCID: PMC2911109
- DOI: 10.1186/gb-2010-11-6-r61
Item in Clipboard
Design and evaluation of genome-wide libraries for RNA interference screens
Genome Biol.
2010.
Abstract
RNA interference (RNAi) screens have enabled the systematic analysis of many biological processes in cultured cells and whole organisms. The success of such screens and the interpretation of the data depend on the stringent design of RNAi libraries. We describe and validate NEXT-RNAi, a software for the automated design and evaluation of RNAi sequences on a genome-wide scale. NEXT-RNAi is implemented as open-source software and is accessible at http://www.nextrnai.org/.
Figures
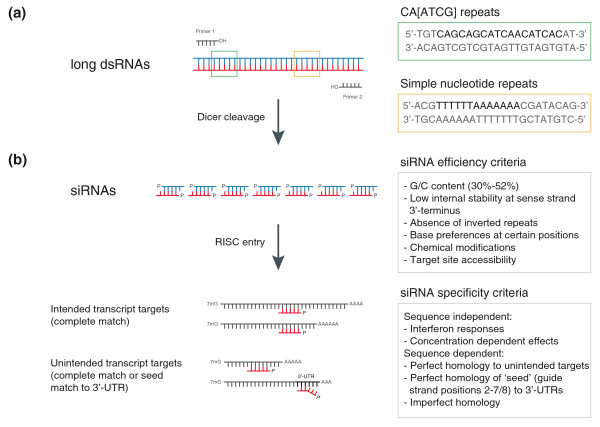
Quality control parameters for RNAi reagents at different stages of the design pipeline. (a) Long dsRNAs that have regions of low complexity, for example, CA[ATCG] repeats or simple nucleotide repeats, can exert unspecific and cytotoxic effects. The quality of the primer designs used to synthesize amplicons from DNA sources is crucial, in particular when the synthesis is performed in 96- or 384-well formats where primers should have similar melting temperatures. (b) Dicer-mediated cleavage of long dsRNAs leads to the generation of siRNAs of lengths between 19 and 23 nucleotides [29]. The quality of siRNAs depends on their ability to efficiently enter the RNA-induced silencing complex (RISC) and to access the target mRNA. This is influenced by thermodynamic properties, base preferences and chemical modifications. The specificity of siRNAs is influenced by sequence-independent and sequence-dependent features. siRNAs can trigger interferon responses or show concentration-dependent cytotoxic effects, independent of their sequence. Silencing of unintended target transcripts can occur through perfect and imperfect sequence homologies to the siRNA and through 'seed matches' to the transcript 3' UTRs. See text for details.
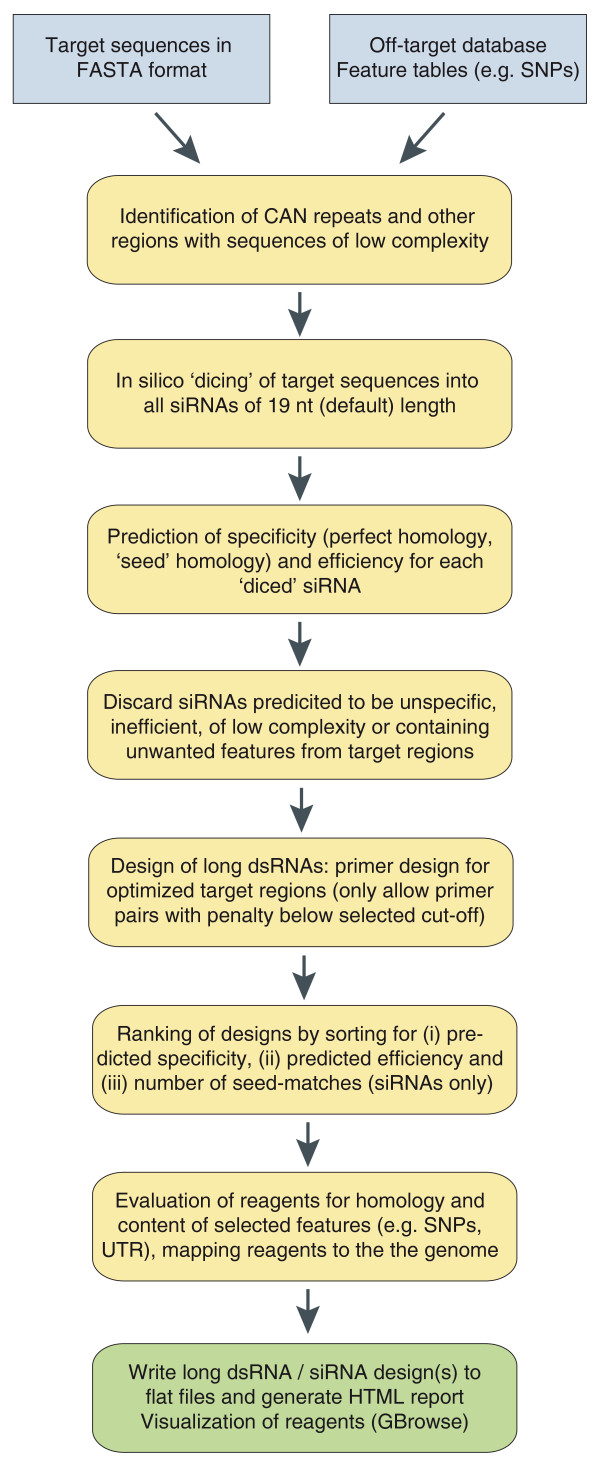
Overview of the NEXT-RNAi workflow. NEXT-RNAi requires a defined set of input files in FASTA or tab-delimited formats. First, the program filters the input target sequences for six (default) or more contiguous CAN repeats and for other regions of low complexity (for example, simple nucleotide repeats) using mdust. Sequences are then 'diced' to generate all possible siRNA sequences with a default length of 19 nucleotides (nt) and an offset of 1 nucleotide. Subsequently, each siRNA is mapped to a user-defined off-target database (for example, the whole transcriptome) with Bowtie [37] to determine its specificity. The specificity is set to one if the siRNA targets a single gene or to zero otherwise. In the next step, the predicted efficiency of each 19-nucleotide siRNA is computed. Two methods can be selected, the 'rational' method according to Reynolds et al. [9] and the 'weighted' method according to Shah et al. [12], assigning each siRNA an efficiency score between 0 and 100. Optionally, the seed complement frequency for each siRNA can be computed for any FASTA file provided (for example, a file containing 3' UTR sequences). siRNAs that did not pass the low-complexity filters, show perfect homology to multiple target genes or do not meet the user-defined cutoffs for efficiency or seed complement frequency are excluded from the queried target sequences. Remaining sequences are used as templates for primer design (with Primer3 [36]) for long dsRNAs or are directly subjected to the final ranking for the design of siRNAs. Designs are ranked by (i) their predicted specificity and (ii) their predicted efficiency and, in the case of siRNA designs, (iii) their calculated seed complement frequency. Sequences can also be evaluated for additional features, such as homology to unintended transcripts, or SNP and UTR contents. Final designs can be visualized using GBrowse [40]. All results are presented in a comprehensive HTML report and are also exported to text files.
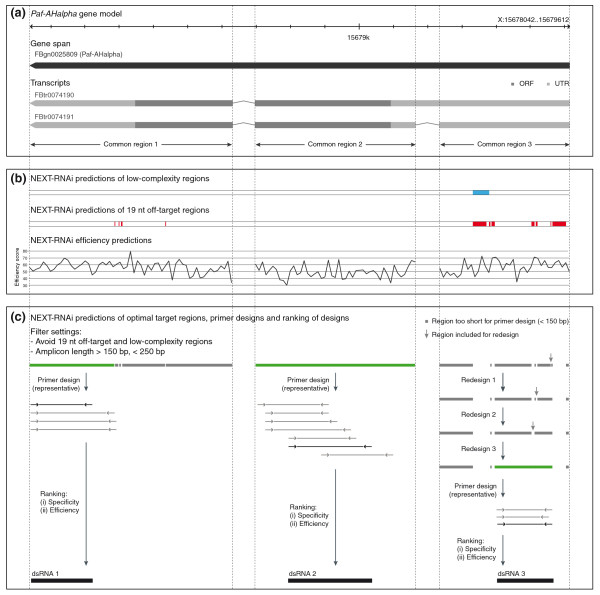
Example of design and filter methods applied by NEXT-RNAi. (a) Visualization of the Paf-AHalpha gene model and transcripts. Regions labeled as 'common region' serve as input for NEXT-RNAi (ORF = open reading frame, UTR = untranslated region). (b) Quality measures computed by NEXT-RNAi for the common regions. Blue and red regions label predicted low-complexity regions (including CAN repeats) and 19-nucleotide off-target regions, respectively. The lower panel shows the predicted siRNA efficiency according to Shah et al. [12] (averaged for ten siRNAs). (c) NEXT-RNAi predictions of optimal target sites (green) after discarding 19-nucleotide off-target and low complexity regions and regions <150 nucleotides or >250 nucleotides. If available, these regions are directly used as templates for primer designs (left and middle panels). Otherwise, a redesign method is used that connects closest 'optimal' neighbors until a region suitable for primer designs is identified (right panel). Potential dsRNAs are finally ranked by their specificity and efficiency.
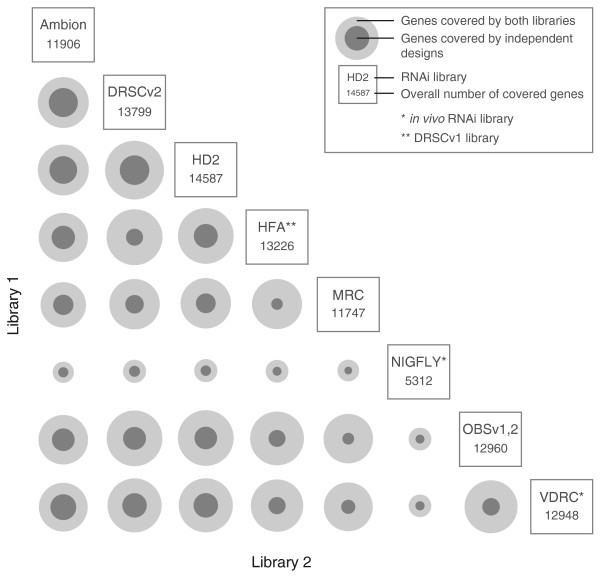
Pairwise comparison of Drosophila RNAi libraries. Eight Drosophila RNAi libraries are compared to identify the number of genes that are targeted by multiple libraries (outer ring, light grey) and the number of genes that are targeted by independent designs (inner ring, dark grey). The reference for the ring sizes is the combination of libraries commonly targeting the most genes (HD2/DRSCv2: 13,197 genes).
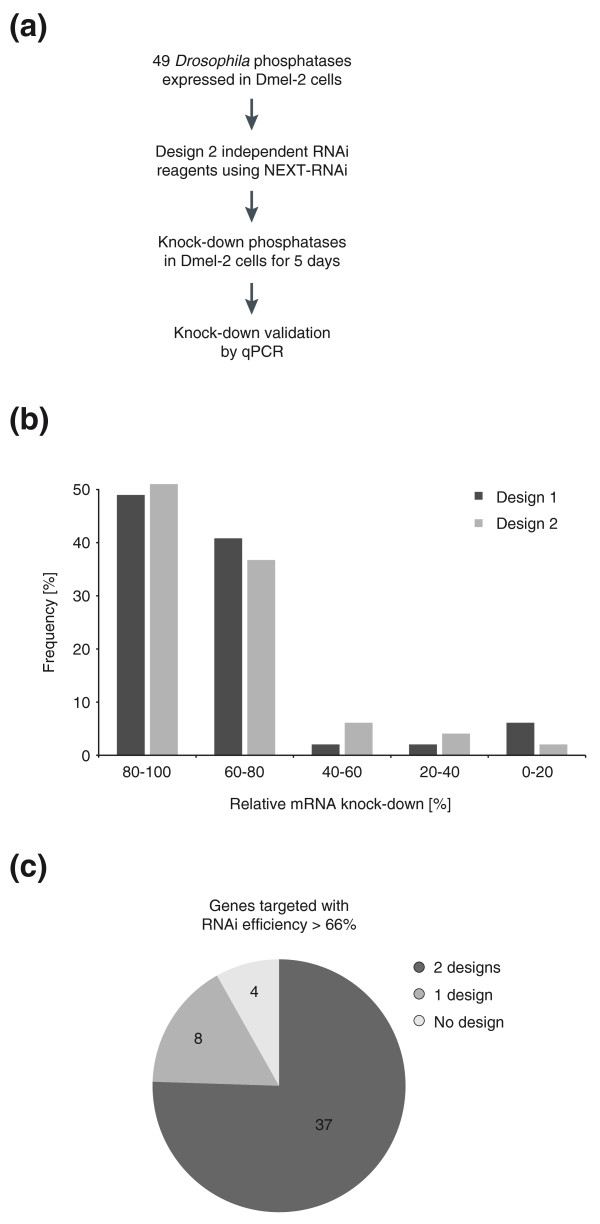
Knock-down validation of Drosophila phosphatases. (a) Experimental workflow for knock-down validation. (b) Frequencies of observed knock-down efficiencies for independent designs. (c) The number of genes efficiently silenced (knock-down >66%) by both independent designs, only one or neither design. qPCR, quantitative RT-PCR.
Similar articles
-
A Guide to Genome-Wide In Vivo RNAi Applications in Drosophila.Methods Mol Biol. 2016;1478:117-143. doi: 10.1007/978-1-4939-6371-3_6. Methods Mol Biol. 2016. PMID: 27730578 Review.
-
E-RNAi: a web application to design optimized RNAi constructs.Nucleic Acids Res. 2005 Jul 1;33(Web Server issue):W582-8. doi: 10.1093/nar/gki468. Nucleic Acids Res. 2005. PMID: 15980541 Free PMC article.
-
GenomeRNAi: a database for cell-based RNAi phenotypes. 2009 update.Nucleic Acids Res. 2010 Jan;38(Database issue):D448-52. doi: 10.1093/nar/gkp1038. Epub 2009 Nov 12. Nucleic Acids Res. 2010. PMID: 19910367 Free PMC article.
-
RNA Interference (RNAi) Screening in Drosophila.Genetics. 2018 Mar;208(3):853-874. doi: 10.1534/genetics.117.300077. Genetics. 2018. PMID: 29487145 Free PMC article.
-
Genome-wide RNAi as a route to gene function in Drosophila.Brief Funct Genomic Proteomic. 2004 Aug;3(2):168-76. doi: 10.1093/bfgp/3.2.168. Brief Funct Genomic Proteomic. 2004. PMID: 15355598 Review.
Cited by
-
Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy.Proc Natl Acad Sci U S A. 2014 Jun 10;111(23):8494-9. doi: 10.1073/pnas.1321207111. Epub 2014 May 27. Proc Natl Acad Sci U S A. 2014. PMID: 24912190 Free PMC article.
-
Advances in genome-wide RNAi cellular screens: a case study using the Drosophila JAK/STAT pathway.BMC Genomics. 2012 Sep 24;13:506. doi: 10.1186/1471-2164-13-506. BMC Genomics. 2012. PMID: 23006893 Free PMC article.
-
E-TALEN: a web tool to design TALENs for genome engineering.Nucleic Acids Res. 2013 Nov;41(20):e190. doi: 10.1093/nar/gkt789. Epub 2013 Sep 3. Nucleic Acids Res. 2013. PMID: 24003033 Free PMC article.
-
Advances in RNAi-Assisted Strain Engineering in Saccharomyces cerevisiae.Front Bioeng Biotechnol. 2020 Jul 2;8:731. doi: 10.3389/fbioe.2020.00731. eCollection 2020. Front Bioeng Biotechnol. 2020. PMID: 32714914 Free PMC article. Review.
-
A genome-wide RNAi screen for genes important for proliferation of cultured Drosophila cells at low temperature identifies the Ball/VRK protein kinase.Chromosoma. 2023 Mar;132(1):31-53. doi: 10.1007/s00412-023-00787-6. Epub 2023 Feb 7. Chromosoma. 2023. PMID: 36746786 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
