

Detecting lateral gene transfers by statistical reconciliation of phylogenetic forests
- PMID: 20550700
- PMCID: PMC2905365
- DOI: 10.1186/1471-2105-11-324
Detecting lateral gene transfers by statistical reconciliation of phylogenetic forests
Abstract
Background: To understand the evolutionary role of Lateral Gene Transfer (LGT), accurate methods are needed to identify transferred genes and infer their timing of acquisition. Phylogenetic methods are particularly promising for this purpose, but the reconciliation of a gene tree with a reference (species) tree is computationally hard. In addition, the application of these methods to real data raises the problem of sorting out real and artifactual phylogenetic conflict.
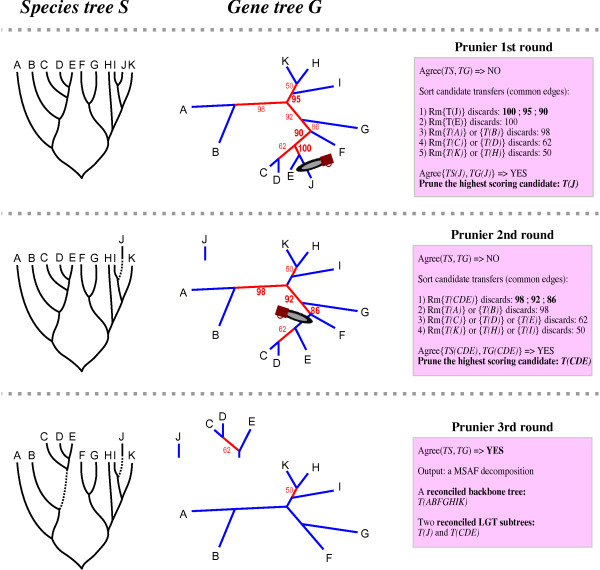
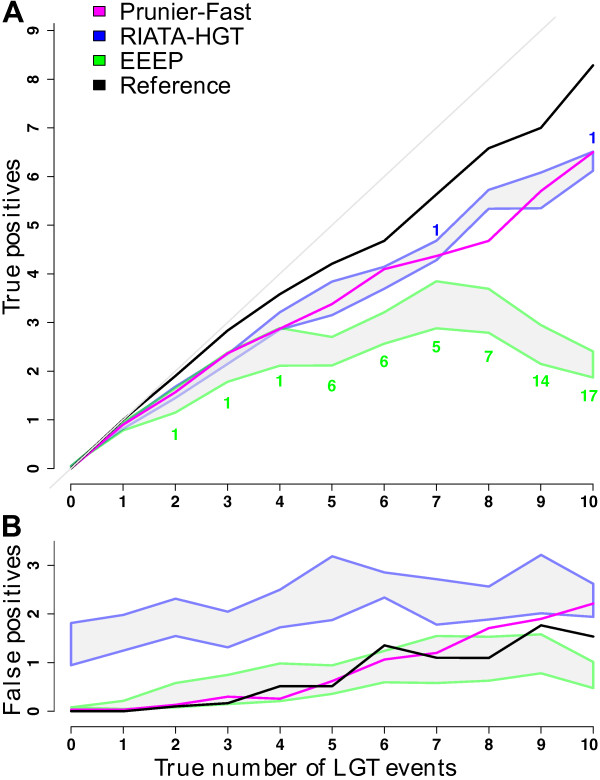
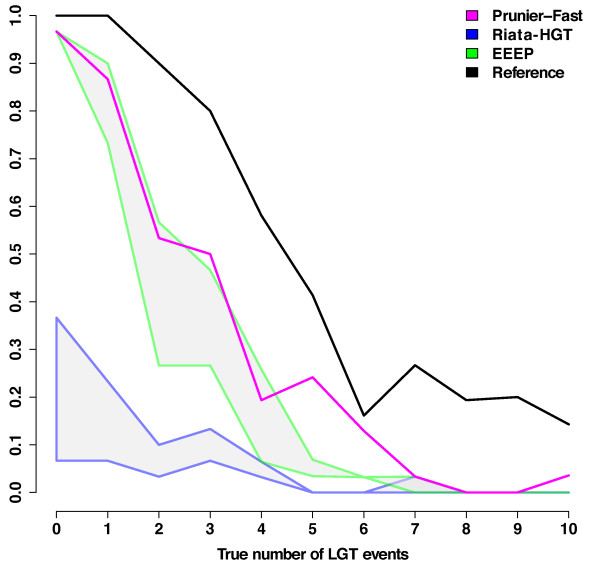
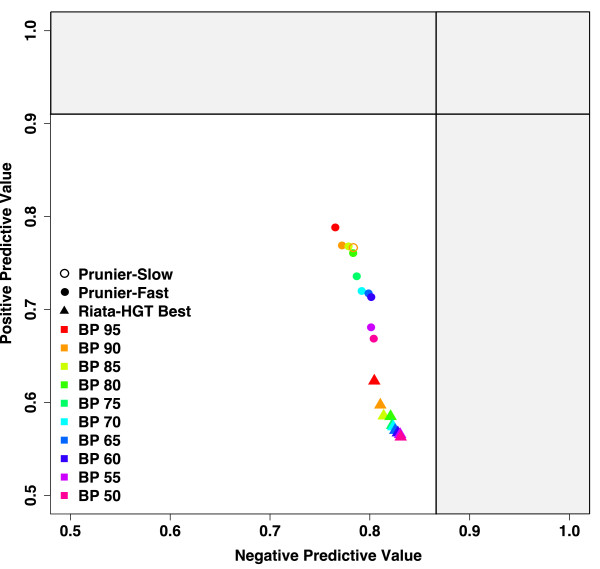
Results: We present Prunier, a new method for phylogenetic detection of LGT based on the search for a maximum statistical agreement forest (MSAF) between a gene tree and a reference tree. The program is flexible as it can use any definition of "agreement" among trees. We evaluate the performance of Prunier and two other programs (EEEP and RIATA-HGT) for their ability to detect transferred genes in realistic simulations where gene trees are reconstructed from sequences. Prunier proposes a single scenario that compares to the other methods in terms of sensitivity, but shows higher specificity. We show that LGT scenarios carry a strong signal about the position of the root of the species tree and could be used to identify the direction of evolutionary time on the species tree. We use Prunier on a biological dataset of 23 universal proteins and discuss their suitability for inferring the tree of life.
Conclusions: The ability of Prunier to take into account branch support in the process of reconciliation allows a gain in complexity, in comparison to EEEP, and in accuracy in comparison to RIATA-HGT. Prunier's greedy algorithm proposes a single scenario of LGT for a gene family, but its quality always compares to the best solutions provided by the other algorithms. When the root position is uncertain in the species tree, Prunier is able to infer a scenario per root at a limited additional computational cost and can easily run on large datasets.Prunier is implemented in C++, using the Bio++ library and the phylogeny program Treefinder. It is available at: http://pbil.univ-lyon1.fr/software/prunier.
Figures
References
-
- Gogarten JP, Doolittle WF, Lawrence JG. Prokaryotic evolution in light of gene transfer. Mol Biol Evol. 2002;19:2226–2238. - PubMed
