

Essential requirement for PP2A inhibition by the oncogenic receptor c-KIT suggests PP2A reactivation as a strategy to treat c-KIT+ cancers
- PMID: 20551067
- PMCID: PMC2933456
- DOI: 10.1158/0008-5472.CAN-09-2544
Essential requirement for PP2A inhibition by the oncogenic receptor c-KIT suggests PP2A reactivation as a strategy to treat c-KIT+ cancers
Abstract
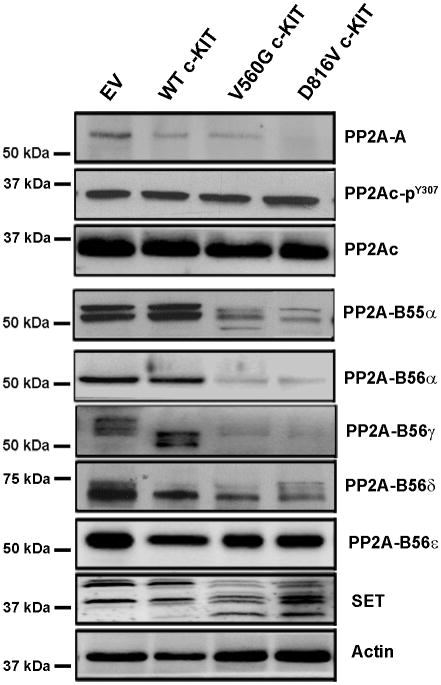
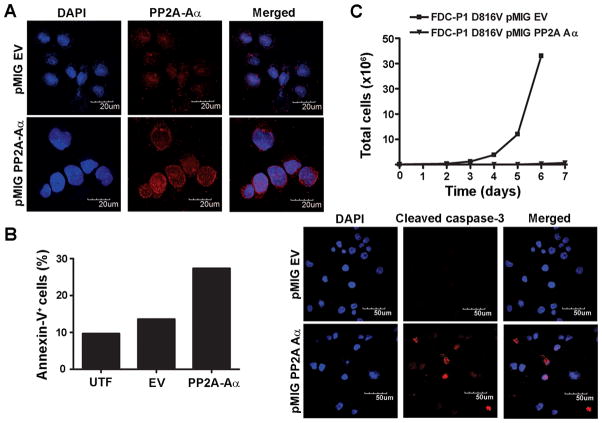
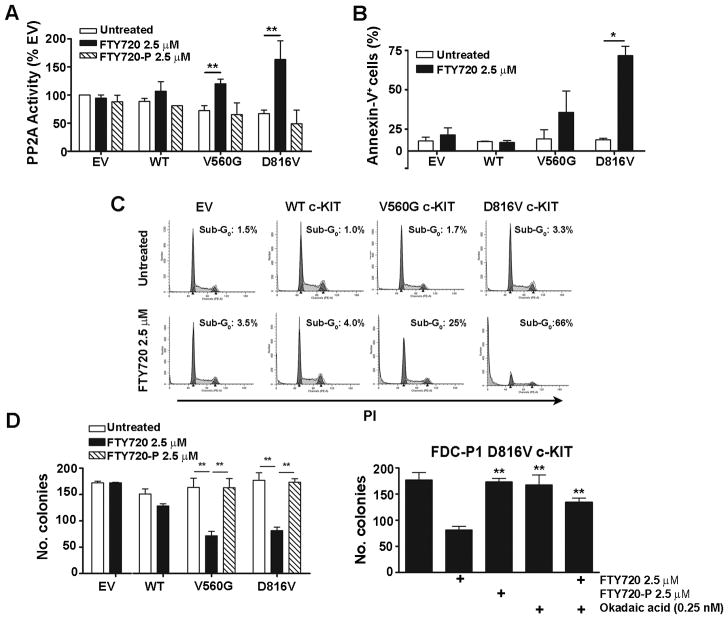
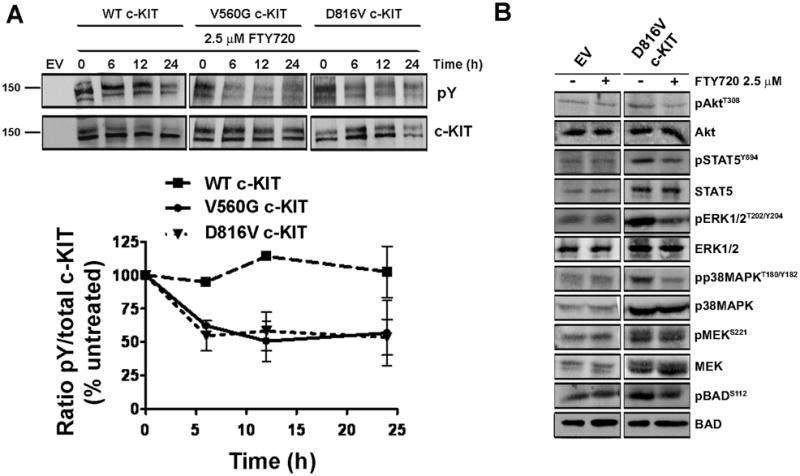
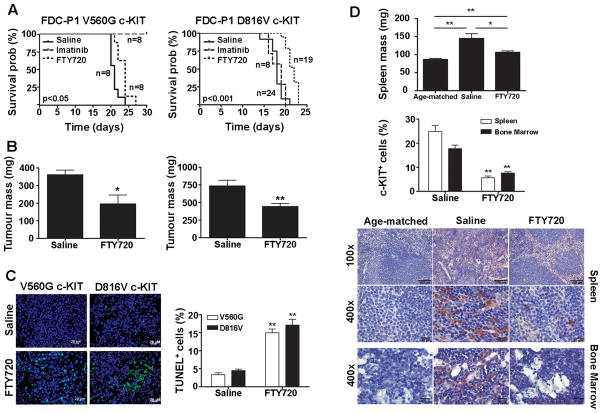
Oncogenic mutations of the receptor tyrosine kinase c-KIT play an important role in the pathogenesis of gastrointestinal stromal tumors, systemic mastocytosis, and some acute myeloid leukemias (AML). Although juxtamembrane mutations commonly detected in gastrointestinal stromal tumor are sensitive to tyrosine kinase inhibitors, the kinase domain mutations frequently encountered in systemic mastocytosis and AML confer resistance and are largely unresponsive to targeted inhibition by the existing agent imatinib. In this study, we show that myeloid cells expressing activated c-KIT mutants that are imatinib sensitive (V560G) or imatinib resistant (D816V) can inhibit the tumor suppressor activity of protein phosphatase 2A (PP2A). This effect was associated with the reduced expression of PP2A structural (A) and regulatory subunits (B55alpha, B56alpha, B56gamma, and B56delta). Overexpression of PP2A-Aalpha in D816V c-KIT cells induced apoptosis and inhibited proliferation. In addition, pharmacologic activation of PP2A by FTY720 reduced proliferation, inhibited clonogenic potential, and induced apoptosis of mutant c-KIT(+) cells, while having no effect on wild-type c-KIT cells or empty vector controls. FTY720 treatment caused the dephosphorylation of the D816V c-KIT receptor and its downstream signaling targets pAkt, pSTAT5, and pERK1/2. Additionally, in vivo administration of FTY720 delayed the growth of V560G and D816V c-KIT tumors, inhibited splenic and bone marrow infiltration, and prolonged survival. Our findings show that PP2A inhibition is essential for c-KIT-mediated tumorigenesis, and that reactivating PP2A may offer an attractive strategy to treat drug-resistant c-KIT(+) cancers.
Copyright 2010 AACR.
Conflict of interest statement
The authors declare no additional competing financial interests.
Figures
Comment in
-
Essential requirement for PP2A inhibition by the oncogenic receptor c-KIT suggests PP2A reactivation as a strategy to treat c-KIT+ cancers -- Letter.Cancer Res. 2011 Mar 15;71(6):2403; author reply 2404. doi: 10.1158/0008-5472.CAN-10-3383. Cancer Res. 2011. PMID: 21406406 No abstract available.
References
-
- Lennartsson J, Jelacic T, Linnekin D, Shivakrupa R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells. 2005;23:16–43. - PubMed
-
- Linnekin D. Early signaling pathways activated by c-Kit in hematopoietic cells. Int J Biochem Cell Biol. 1999;31:1053–74. - PubMed
-
- Paschka P, Marcucci G, Ruppert AS, et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. J Clin Oncol. 2006;24:3904–11. - PubMed
-
- Worobec AS, Semere T, Nagata H, Metcalfe DD. Clinical correlates of the presence of the Asp816Val c-kit mutation in the peripheral blood mononuclear cells of patients with mastocytosis. Cancer. 1998;83:2120–9. - PubMed
-
- Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–80. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
