

Understanding resistance to EGFR inhibitors-impact on future treatment strategies
- PMID: 20551942
- PMCID: PMC2929287
- DOI: 10.1038/nrclinonc.2010.97
Understanding resistance to EGFR inhibitors-impact on future treatment strategies
Abstract
EGFR is a tyrosine kinase that participates in the regulation of cellular homeostasis. Following ligand binding, EGFR stimulates downstream cell signaling cascades that influence cell proliferation, apoptosis, migration, survival and complex processes, including angiogenesis and tumorigenesis. EGFR has been strongly implicated in the biology of human epithelial malignancies, with therapeutic applications in cancers of the colon, head and neck, lung, and pancreas. Accordingly, targeting EGFR has been intensely pursued, with the development of a series of promising molecular inhibitors for use in clinical oncology. As is common in cancer therapy, challenges with respect to treatment resistance emerge over time. This situation is certainly true of EGFR inhibitor therapies, where intrinsic and acquired resistance is now well recognized. In this Review, we provide a brief overview regarding the biology of EGFR, preclinical and clinical development of EGFR inhibitors, and molecular mechanisms that underlie the development of treatment resistance. A greater understanding of the mechanisms that lead to EGFR resistance may provide valuable insights to help design new strategies that will enhance the impact of this promising class of inhibitors for the treatment of cancer.
Figures
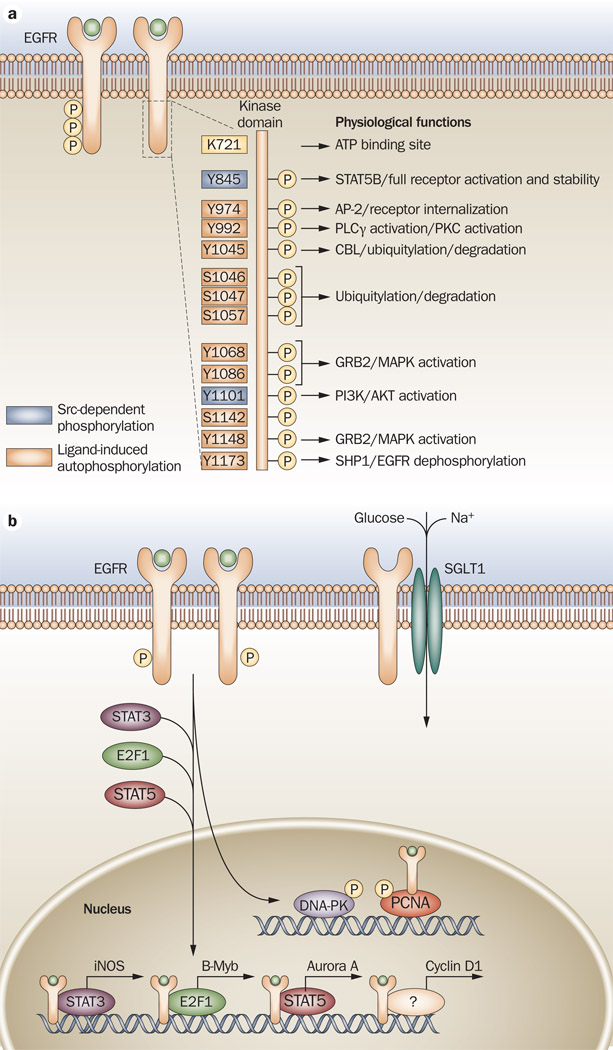
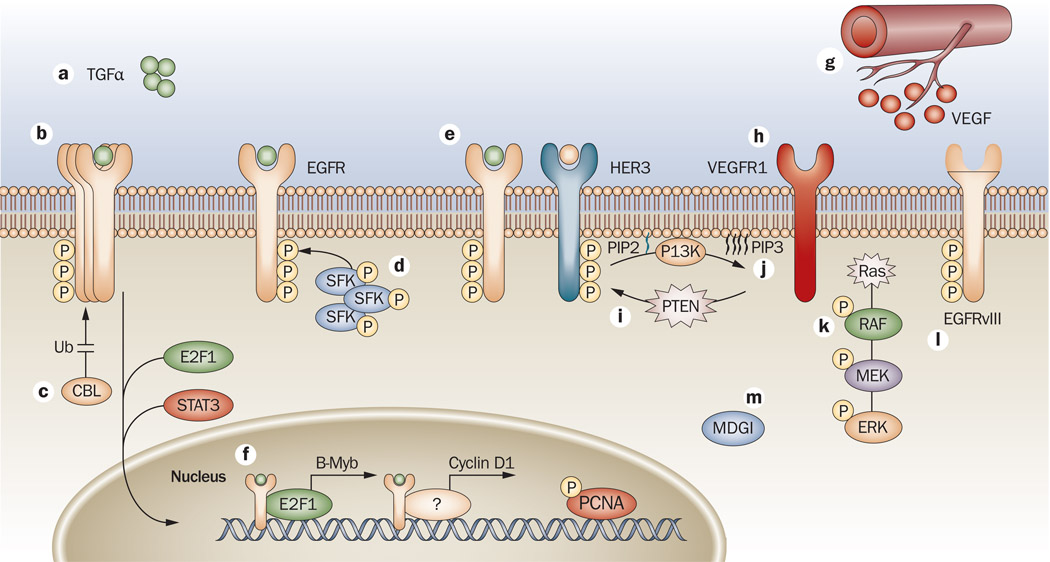
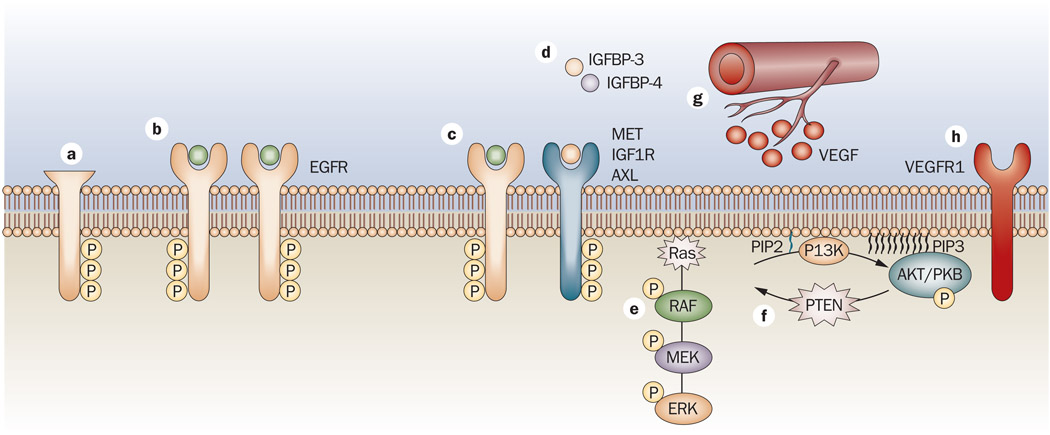
References
-
- Cohen S. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J. Biol. Chem. 1962;237:1555–1562. - PubMed
-
- Cohen S. The stimulation of epidermal proliferation by a specific protein (EGF) Dev. Biol. 1965;12:394–407. - PubMed
-
- Carpenter G, Lembach KJ, Morrison MM, Cohen S. Characterization of the binding of 125-I-labeled epidermal growth factor to human fibroblasts. J. Biol. Chem. 1975;250:4297–4304. - PubMed
-
- Carpenter G, King L, Jr, Cohen S. Epidermal growth factor stimulates phosphorylation in membrane preparations in vitro. Nature. 1978;276:409–410. - PubMed
-
- Ullrich A, et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature. 1984;309:418–425. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
