

Compound deficiencies in multiple fibroblast growth factor signalling components differentially impact the murine gonadotrophin-releasing hormone system
- PMID: 20553372
- PMCID: PMC3102046
- DOI: 10.1111/j.1365-2826.2010.02024.x
Compound deficiencies in multiple fibroblast growth factor signalling components differentially impact the murine gonadotrophin-releasing hormone system
Abstract
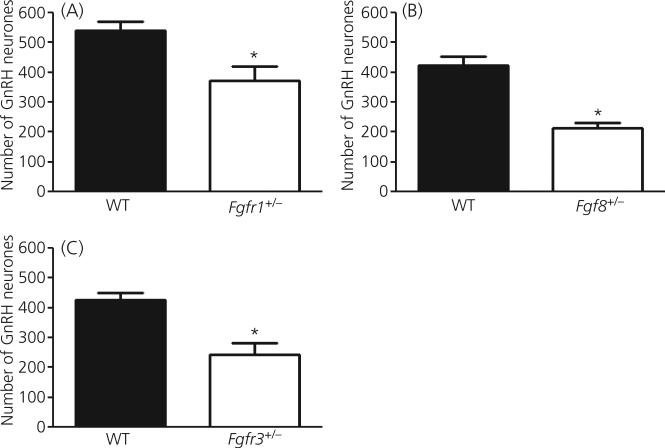
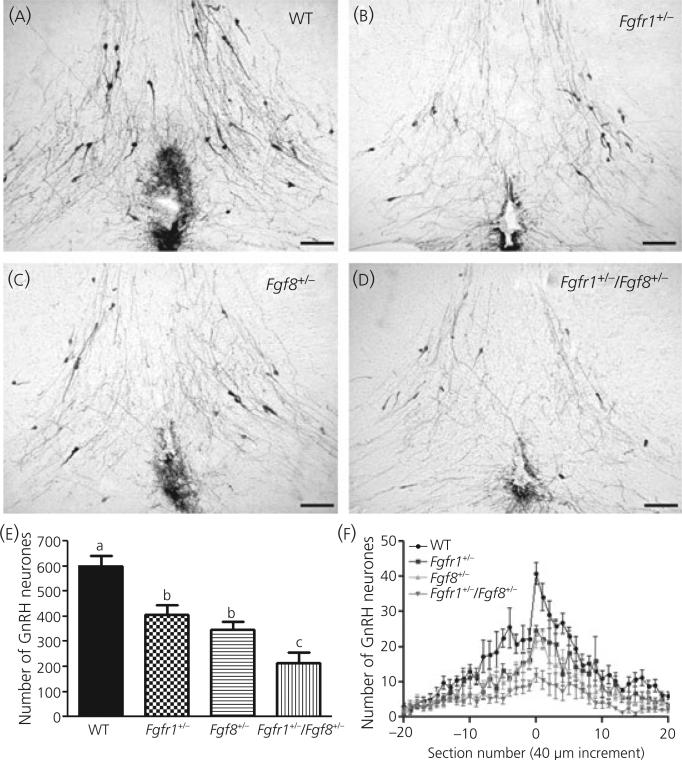
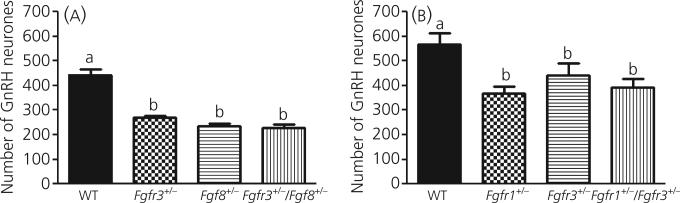
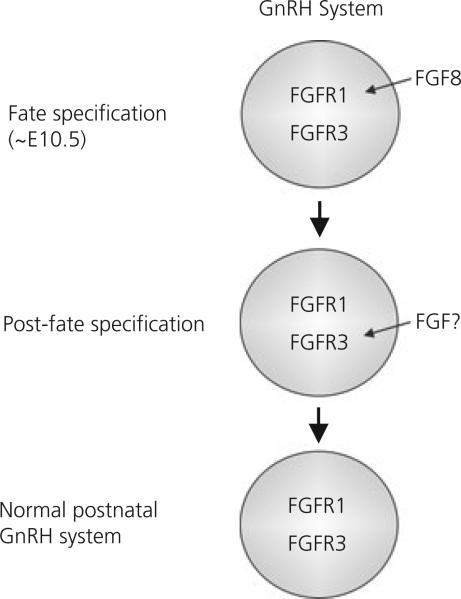
Gonadotrophin-releasing hormone (GnRH) neurones control the onset and maintenance of fertility. Aberrant development of the GnRH system underlies infertility in Kallmann syndrome [KS; idiopathic hypogonadotropic hypogonadism (IHH) and anosmia]. Some KS patients harbour mutations in the fibroblast growth factor receptor 1 (Fgfr1) and Fgf8 genes. The biological significance of these two genes in GnRH neuronal development was corroborated by the observation that GnRH neurones were severely reduced in newborn transgenic mice deficient in either gene. In the present study, we hypothesised that the compound deficiency of Fgf8 and its cognate receptors, Fgfr1 and Fgfr3, may lead to more deleterious effects on the GnRH system, thereby resulting in a more severe reproductive phenotype in patients harbouring these mutations. This hypothesis was tested by counting the number of GnRH neurones in adult transgenic mice with digenic heterozygous mutations in Fgfr1/Fgf8, Fgfr3/Fgf8 or Fgfr1/Fgfr3. Monogenic heterozygous mutations in Fgfr1, Fgf8 or Fgfr3 caused a 30-50% decrease in the total number of GnRH neurones. Interestingly, mice with digenic mutations in Fgfr1/Fgf8 showed a greater decrease in GnRH neurones compared to mice with a heterozygous defect in the Fgfr1 or Fgf8 alone. This compounding effect was not detected in mice with digenic heterozygous mutations in Fgfr3/Fgf8 or Fgfr1/Fgfr3. These results support the hypothesis that IHH/KS patients with digenic mutations in Fgfr1/Fgf8 may have a further reduction in the GnRH neuronal population compared to patients harbouring monogenic haploid mutations in Fgfr1 or Fgf8. Because only Fgfr1/Fgf8 compound deficiency leads to greater GnRH system defect, this also suggests that these fibroblast growth factor signalling components interact in a highly specific fashion to support GnRH neuronal development.
Figures
References
-
- Livne I, Gibson MJ, Silverman AJ. Biochemical differentiation and intercellular interactions of migratory gonadotropin-releasing hormone (GnRH) cells in the mouse. Dev Biol. 1993;159:643–656. - PubMed
-
- Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature. 1989;338:161–164. - PubMed
-
- Franco B, Guioli S, Pragliola A, Incerti B, Bardoni B, Tonlorenzi R, Carrozzo R, Maestrini E, Pieretti M, Taillon-Miller P, Brown CJ, Willard HF, Lawrence C, Persico MG, Camerino G, Ballabio A. A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. 1991;353:529–536. - PubMed
-
- Dode C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, Kottler ML, Lespinasse J, Lienhardt-Roussie A, Mathieu M, Moerman A, Morgan G, Murat A, Toublanc JE, Wolczynski S, Delpech M, Petit C, Young J, Hardelin JP. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2006;2:e175. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
