

Interaction with polyglutamine aggregates reveals a Q/N-rich domain in TDP-43
- PMID: 20554523
- PMCID: PMC2924052
- DOI: 10.1074/jbc.M110.125039
Interaction with polyglutamine aggregates reveals a Q/N-rich domain in TDP-43
Abstract
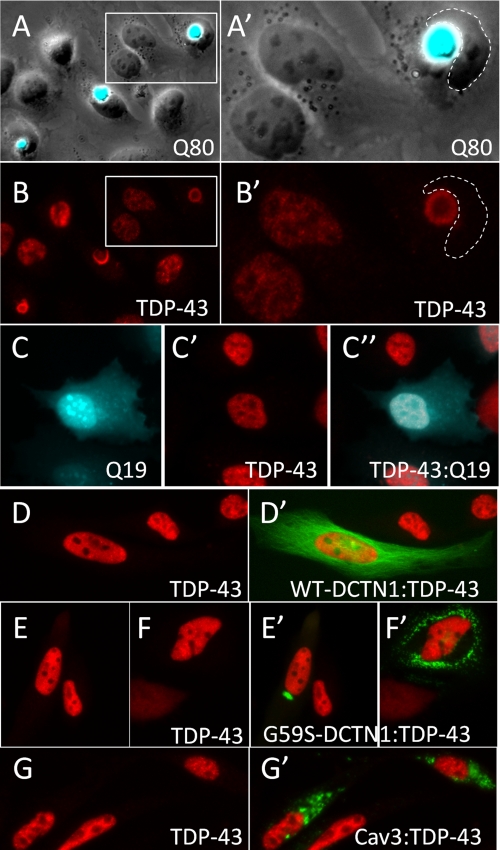
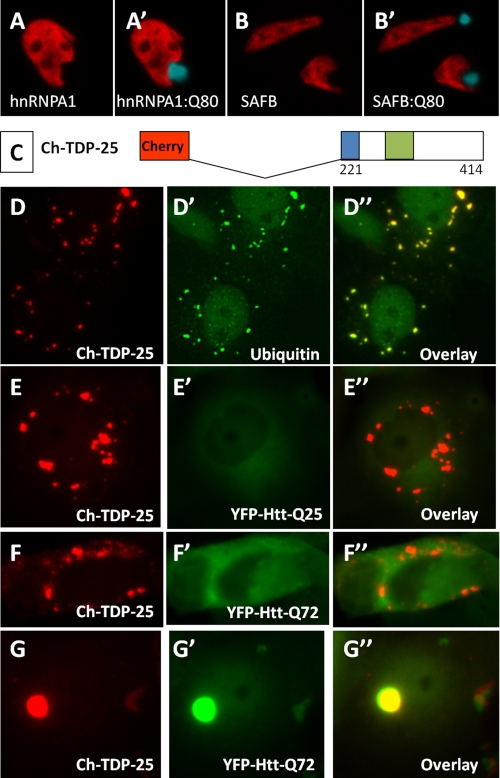
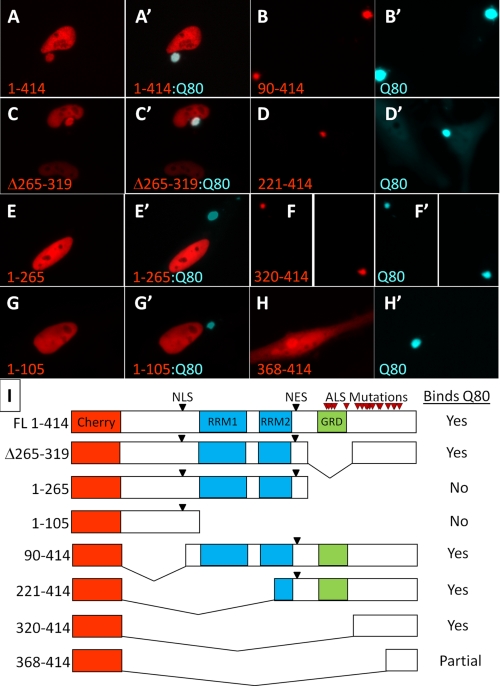
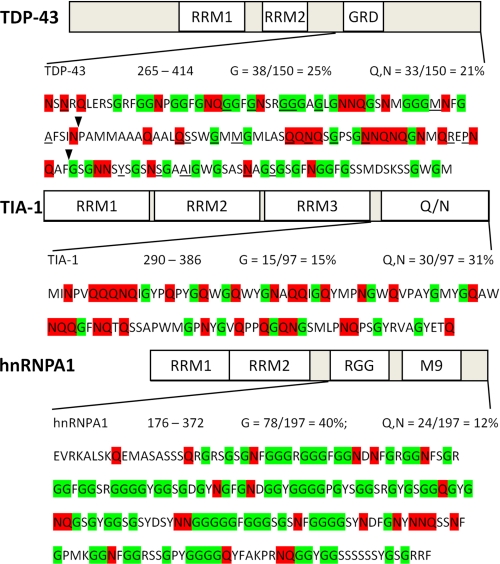
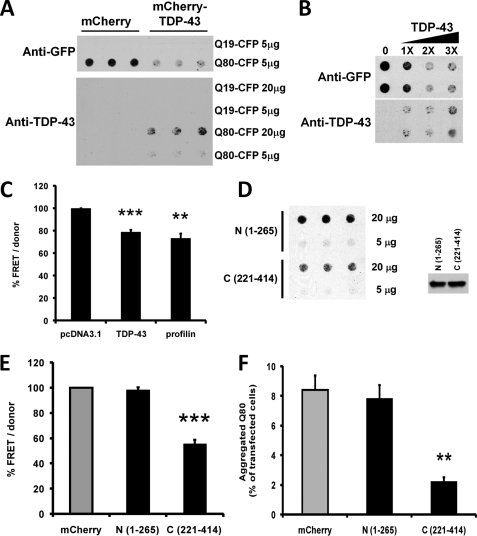
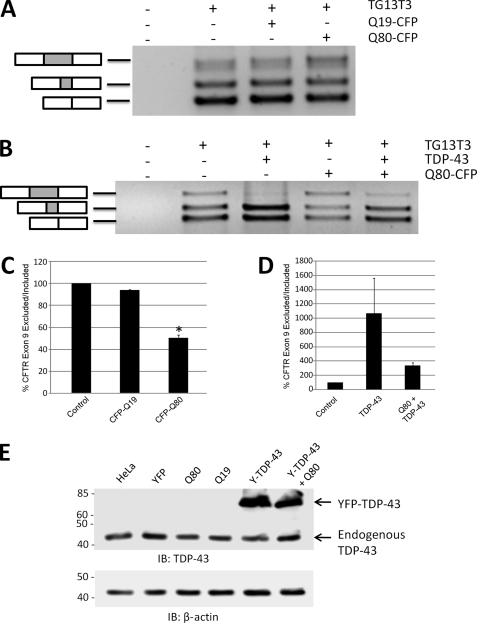
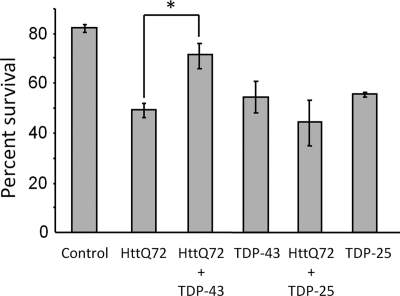
The identification of pathologic TDP-43 aggregates in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration, followed by the discovery of dominantly inherited point mutations in TDP-43 in familial ALS, have been critical insights into the mechanism of these untreatable neurodegenerative diseases. However, the biochemical basis of TDP-43 aggregation and the mechanism of how mutations in TDP-43 lead to disease remain enigmatic. In efforts to understand how TDP-43 alters its cellular localization in response to proteotoxic stress, we found that TDP-43 is sequestered into polyglutamine aggregates. Furthermore, we found that binding to polyglutamine aggregates requires a previously uncharacterized glutamine/asparagine (Q/N)-rich region in the C-terminal domain of TDP-43. Sequestration into polyglutamine aggregates causes TDP-43 to be cleared from the nucleus and become detergent-insoluble. Finally, we observed that sequestration into polyglutamine aggregates led to loss of TDP-43-mediated splicing in the nucleus and that polyglutamine toxicity could be partially rescued by increasing expression of TDP-43. These data indicate pathologic sequestration into polyglutamine aggregates, and loss of nuclear TDP-43 function may play an unexpected role in polyglutamine disease pathogenesis. Furthermore, as Q/N domains have a strong tendency to self-aggregate and in some cases can function as prions, the identification of a Q/N domain in TDP-43 has important implications for the mechanism of pathologic aggregation of TDP-43 in ALS and other neurodegenerative diseases.
Figures
References
-
- Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., McCluskey L. F., Miller B. L., Masliah E., Mackenzie I. R., Feldman H., Feiden W., Kretzschmar H. A., Trojanowski J. Q., Lee V. M. (2006) Science 314, 130–133 - PubMed
-
- Yokoseki A., Shiga A., Tan C. F., Tagawa A., Kaneko H., Koyama A., Eguchi H., Tsujino A., Ikeuchi T., Kakita A., Okamoto K., Nishizawa M., Takahashi H., Onodera O. (2008) Ann. Neurol. 63, 538–542 - PubMed
-
- Van Deerlin V. M., Leverenz J. B., Bekris L. M., Bird T. D., Yuan W., Elman L. B., Clay D., Wood E. M., Chen-Plotkin A. S., Martinez-Lage M., Steinbart E., McCluskey L., Grossman M., Neumann M., Wu I. L., Yang W. S., Kalb R., Galasko D. R., Montine T. J., Trojanowski J. Q., Lee V. M., Schellenberg G. D., Yu C. E. (2008) Lancet Neurol. 7, 409–416 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
