

Kelch-like homologue 9 mutation is associated with an early onset autosomal dominant distal myopathy
- PMID: 20554658
- PMCID: PMC2892937
- DOI: 10.1093/brain/awq108
Kelch-like homologue 9 mutation is associated with an early onset autosomal dominant distal myopathy
Erratum in
-
Corrigendum.Brain. 2020 Jun 1;143(6):e52. doi: 10.1093/brain/awaa065. Brain. 2020. PMID: 32163547 Free PMC article. No abstract available.
Abstract
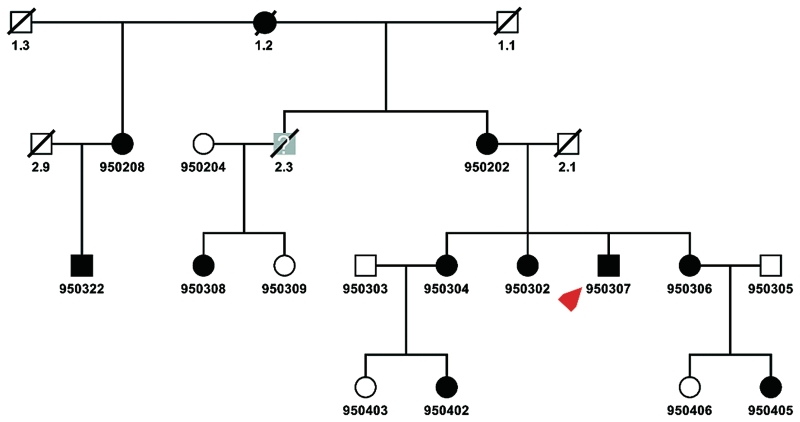
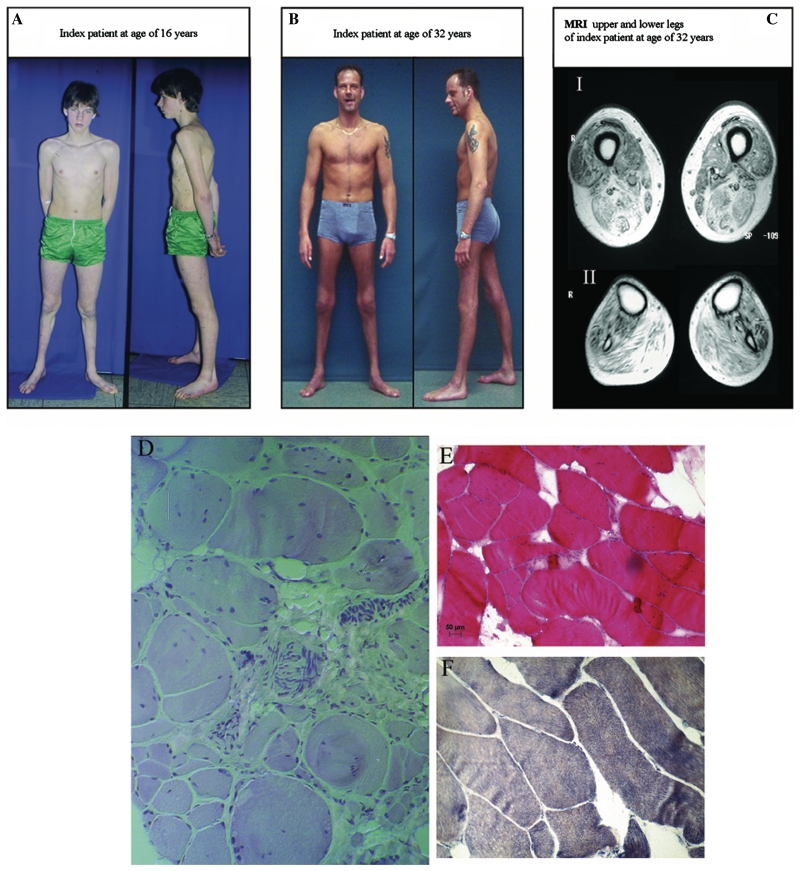
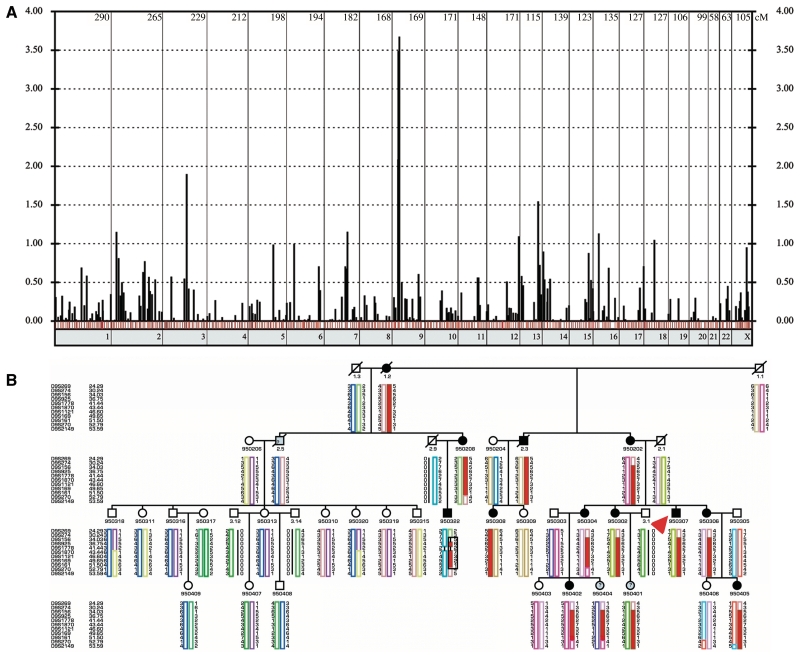
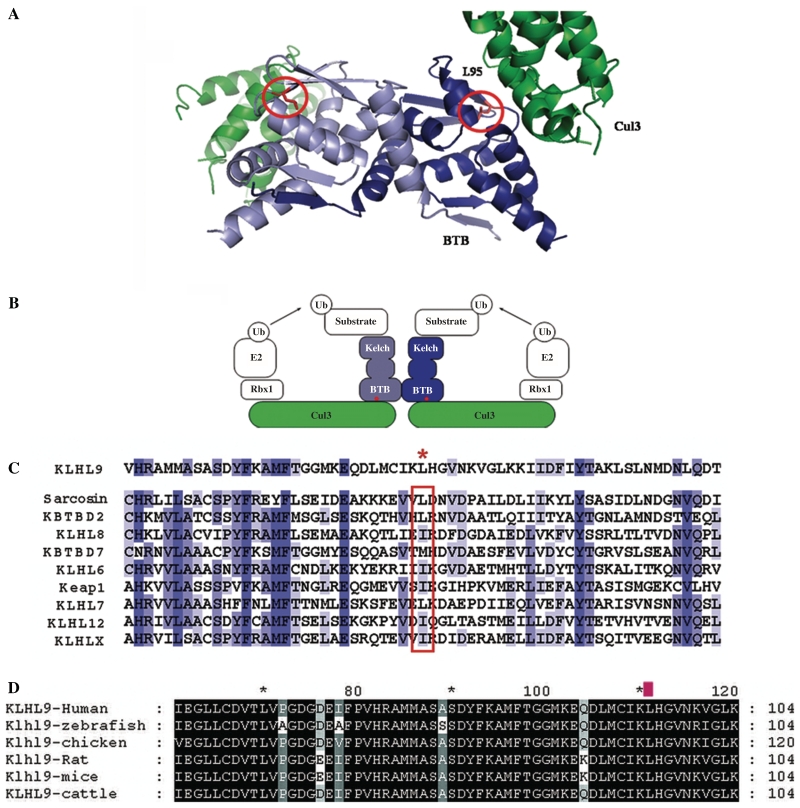
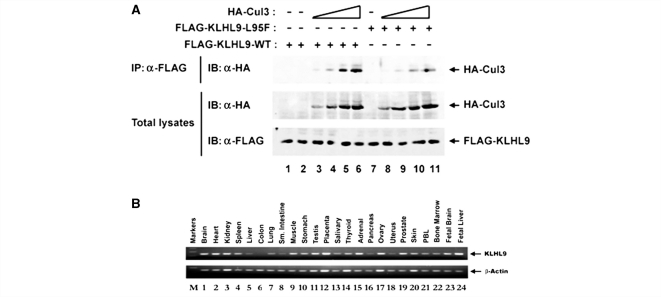
Distal myopathies are a heterogeneous group of disorders characterized by progressive weakness and muscular atrophy, beginning in distal limb muscles and affecting proximal limb muscles at a later stage. We studied a large German kindred with 10 affected members. Weakness and atrophy of the anterior tibial muscles started between the ages of 8 and 16 years, followed by atrophy of intrinsic hand muscles. Progression was slow, and patients retained the ability to walk until the seventh decade. Serum creatinine kinase levels were increased in the range of 150-1400 U/l. Muscle biopsies showed myopathic changes, whereas immunohistochemistry showed normal expression of marker proteins for muscular dystrophies. Patients had reduced sensation with stocking-glove distribution in the distal limbs in later life. Nerve conduction studies revealed no evidence of neuropathy. Genome-wide linkage analysis in this family revealed a new locus for distal myopathy at 9p21.2-p22.3 (multipoint logarithm of the odds ratio=4.21). By positional cloning we found a heterozygous mutation L95F in the Kelch-like homologue 9 gene, encoding a bric-a-brac Kelch protein. Molecular modelling indicated that the mutation may interfere with the interaction of the bric-a-brac domain with Cullin 3. Coimmunoprecipitation experiments confirmed that the mutation reduces association with Cullin 3 in the Kelch-like homologue 9-Cullin 3-E3 ubiquitin ligase complex, which is involved in ubiquitin-dependent protein degradation. We identified a unique form of early onset autosomal dominant distal myopathy which is associated with a Kelch-like homologue 9 mutation and interferes with normal skeletal muscle through a novel pathogenetic mechanism.
Figures
Comment in
-
The age of single-gene neurological disorders is not dead.Brain. 2010 Jul;133(Pt 7):1865-8. doi: 10.1093/brain/awq161. Brain. 2010. PMID: 20584944 Review. No abstract available.
References
-
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–3. - PubMed
-
- Ahlberg G, von Tell D, Borg K, Edstrom L, Anvret M. Genetic linkage of Welander distal myopathy to chromosome 2p13. Ann Neurol. 1999;46:399–404. - PubMed
-
- Aoki M, Arahata K, Brown RH., Jr [Positional cloning of the gene for Miyoshi myopathy and limb-girdle muscular dystrophy] Rinsho Shinkeigaku. 1999;39:1272–5. - PubMed
-
- Bautista J, Rafel E, Castilla JM, Alberca R. Hereditary distal myopathy with onset in early infancy. Observation of a family. J Neurol Sci. 1978;37:149–58. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
