

Fellutamide B is a potent inhibitor of the Mycobacterium tuberculosis proteasome
- PMID: 20558127
- PMCID: PMC2930046
- DOI: 10.1016/j.abb.2010.06.009
Fellutamide B is a potent inhibitor of the Mycobacterium tuberculosis proteasome
Abstract
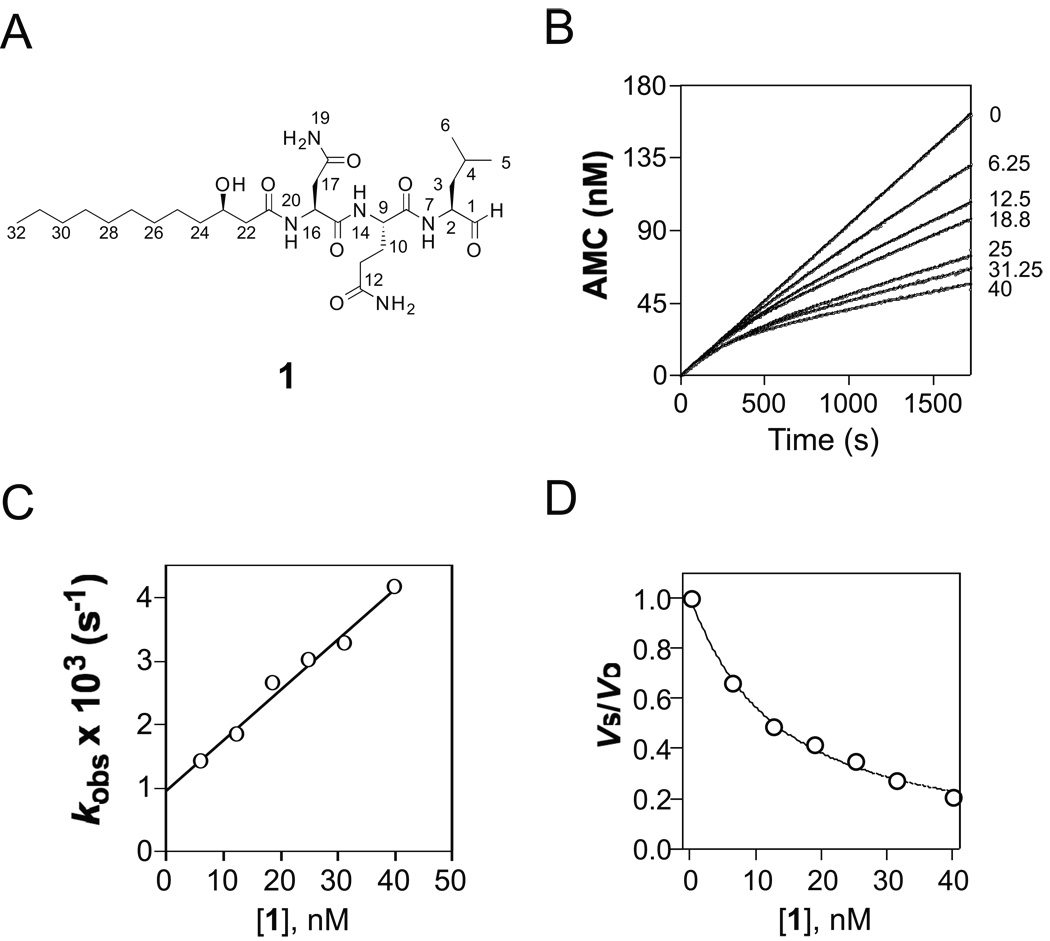
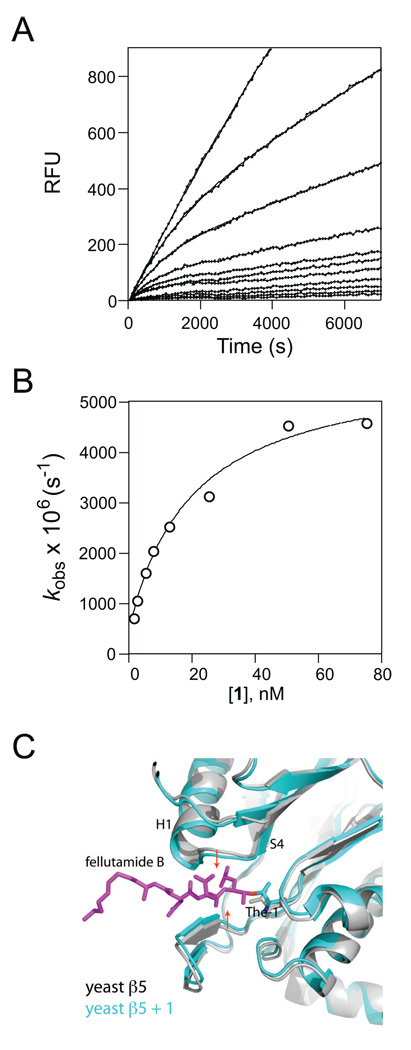
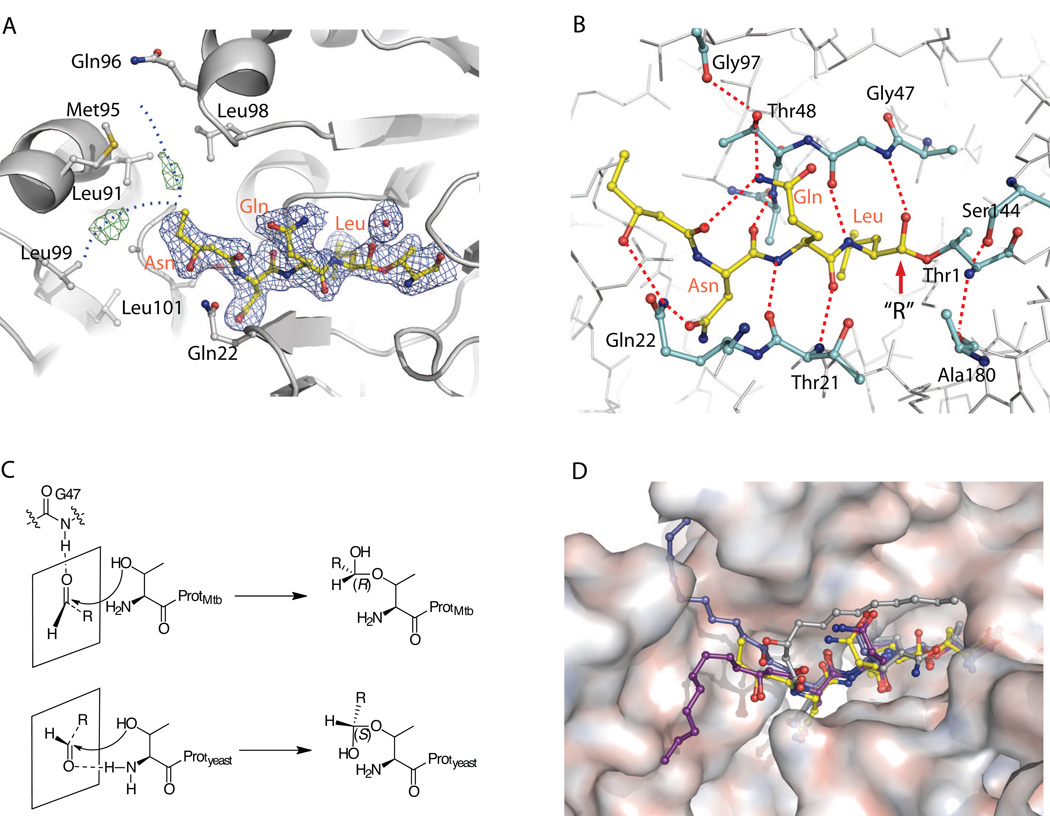
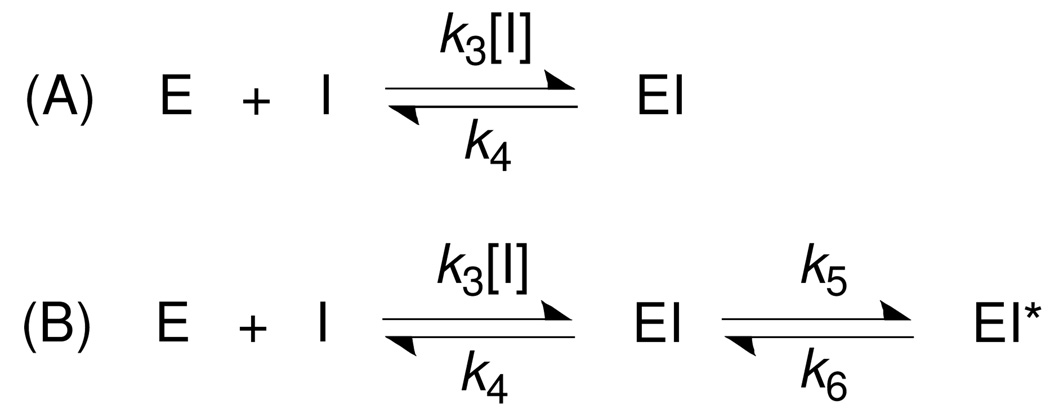
Via high-throughput screening of a natural compound library, we have identified a lipopeptide aldehyde, fellutamide B (1), as the most potent inhibitor of the Mycobacterium tuberculosis (Mtb) proteasome tested to date. Kinetic studies reveal that 1 inhibits both Mtb and human proteasomes in a time-dependent manner under steady-state condition. Remarkably, 1 inhibits the Mtb proteasome in a single-step binding mechanism with K(i)=6.8 nM, whereas it inhibits the human proteasome beta5 active site following a two-step mechanism with K(i)=11.5 nM and K(i)(*)=0.93 nM. Co-crystallization of 1 bound to the Mtb proteasome revealed a structural basis for the tight binding of 1 to the active sites of the Mtb proteasome. The hemiacetal group of 1 in the Mtb proteasome takes the (R)-configuration, whereas in the yeast proteasome it takes the (S)-configuration, indicating that the pre-chiral CHO group of 1 binds to the active site Thr1 in a different orientation. Re-examination of the structure of the yeast proteasome in complex with 1 showed significant conformational changes at the substrate-binding cleft along the active site. These structural differences are consistent with the different kinetic mechanisms of 1 against Mtb and human proteasomes.
Copyright 2010 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Structural Basis for the Species-Selective Binding of N,C-Capped Dipeptides to the Mycobacterium tuberculosis Proteasome.Biochemistry. 2017 Jan 10;56(1):324-333. doi: 10.1021/acs.biochem.6b01107. Epub 2016 Dec 27. Biochemistry. 2017. PMID: 27976853 Free PMC article.
-
Inhibitors selective for mycobacterial versus human proteasomes.Nature. 2009 Oct 1;461(7264):621-6. doi: 10.1038/nature08357. Epub 2009 Sep 16. Nature. 2009. PMID: 19759536 Free PMC article.
-
Mycobacterium tuberculosis prcBA genes encode a gated proteasome with broad oligopeptide specificity.Mol Microbiol. 2006 Mar;59(5):1405-16. doi: 10.1111/j.1365-2958.2005.05035.x. Mol Microbiol. 2006. PMID: 16468985
-
The pup-proteasome system of Mycobacterium tuberculosis.Subcell Biochem. 2013;66:267-95. doi: 10.1007/978-94-007-5940-4_10. Subcell Biochem. 2013. PMID: 23479444 Free PMC article. Review.
-
The Mycobacterium tuberculosis proteasome: more than just a barrel-shaped protease.Microbes Infect. 2009 Dec;11(14-15):1150-5. doi: 10.1016/j.micinf.2009.08.003. Epub 2009 Aug 9. Microbes Infect. 2009. PMID: 19671445 Free PMC article. Review.
Cited by
-
Anti-infective Activity of 2-Cyano-3-Acrylamide Inhibitors with Improved Drug-Like Properties against Two Intracellular Pathogens.Antimicrob Agents Chemother. 2016 Jun 20;60(7):4183-96. doi: 10.1128/AAC.03021-15. Print 2016 Jul. Antimicrob Agents Chemother. 2016. PMID: 27139470 Free PMC article.
-
The mycobacterial proteasomal ATPase Mpa forms a gapped ring to engage the 20S proteasome.J Biol Chem. 2021 Jan-Jun;296:100713. doi: 10.1016/j.jbc.2021.100713. Epub 2021 Apr 27. J Biol Chem. 2021. PMID: 33930464 Free PMC article.
-
Marine-derived Aspergillus species as a source of bioactive secondary metabolites.Mar Biotechnol (NY). 2013 Oct;15(5):499-519. doi: 10.1007/s10126-013-9506-3. Epub 2013 May 25. Mar Biotechnol (NY). 2013. PMID: 23709045 Review.
-
Total Synthesis and Stereochemical Assignment of Nostosin B.Mar Drugs. 2017 Feb 27;15(3):58. doi: 10.3390/md15030058. Mar Drugs. 2017. PMID: 28264450 Free PMC article.
-
Genome mining methods to discover bioactive natural products.Nat Prod Rep. 2021 Nov 17;38(11):2100-2129. doi: 10.1039/d1np00032b. Nat Prod Rep. 2021. PMID: 34734626 Free PMC article. Review.
References
-
- Ryan F. How the battle against tuberculosis was won – and lost. Boston, MA: Lttle, Brown; 1993. The Forgotten Plague.
-
- Murray JF. Am J Respir Crit Care Med. 2004;169:1181–1186. - PubMed
-
- Young D, Hussell T, Dougan G. Nat Immunol. 2002;3:1026–1032. - PubMed
-
- Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. Science. 2003;302:1963–1966. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
