

Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism
- PMID: 20558530
- PMCID: PMC2936746
- DOI: 10.1194/jlr.R005959
Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism
Abstract
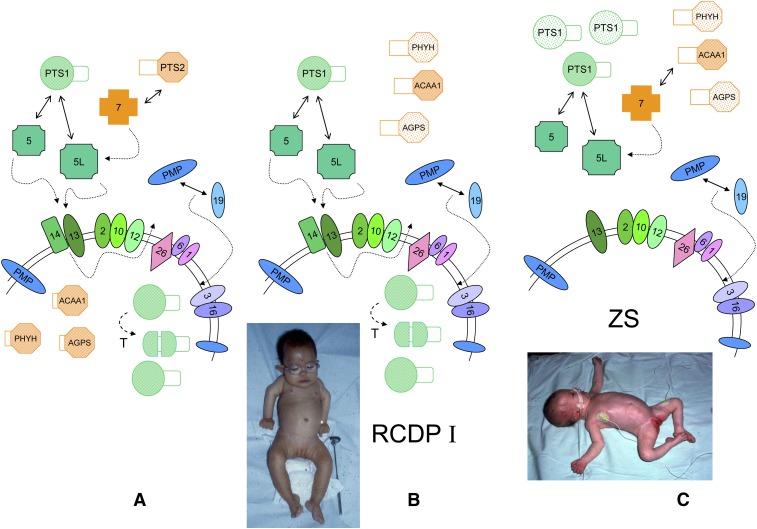
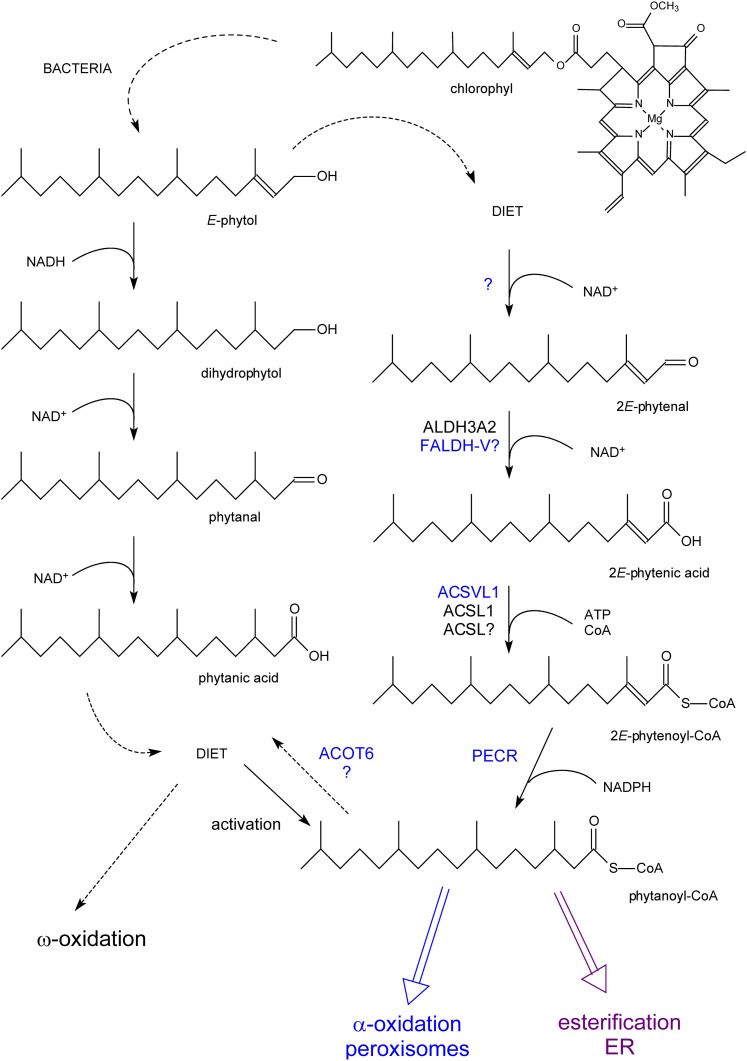
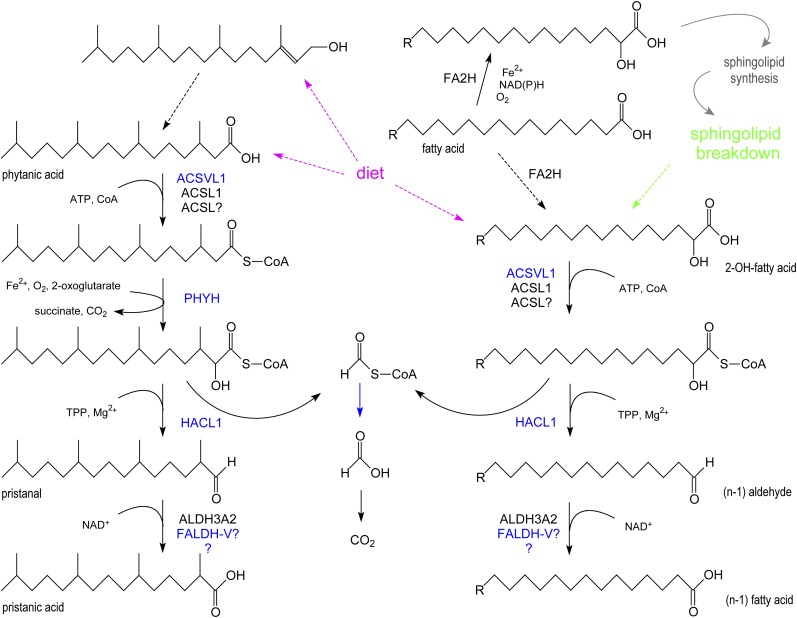
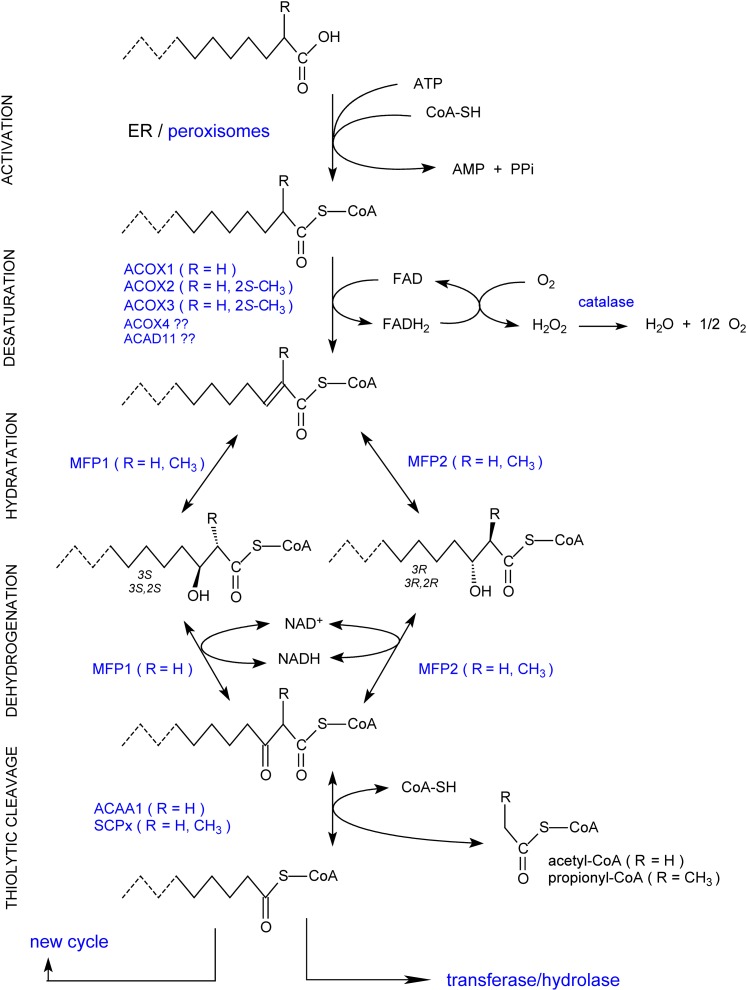
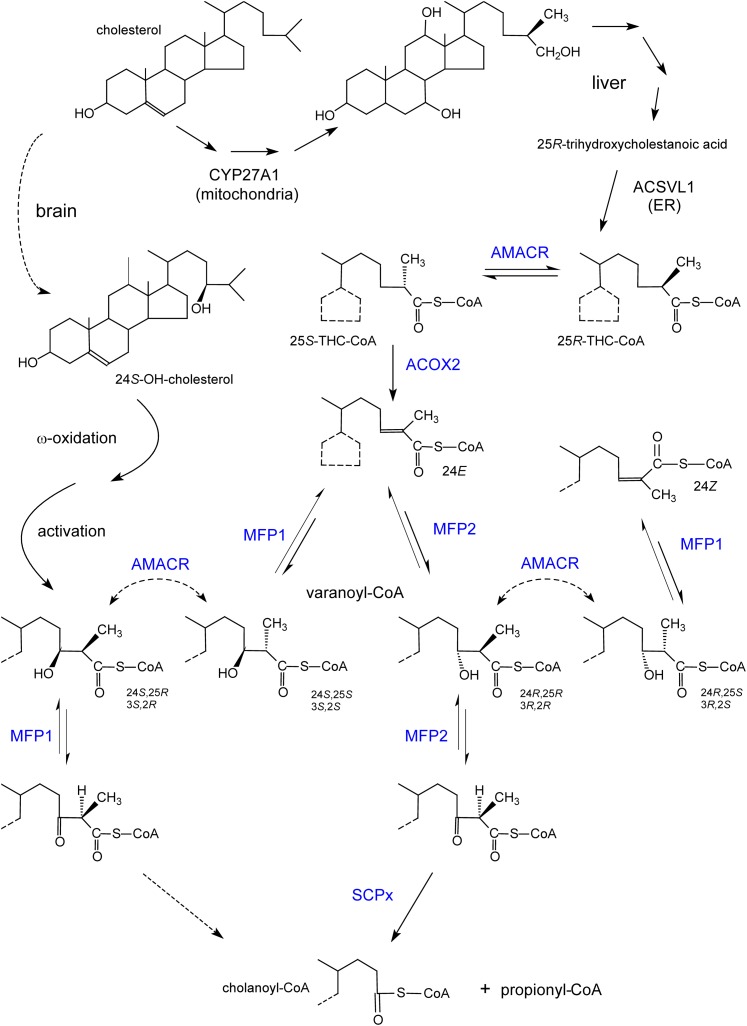
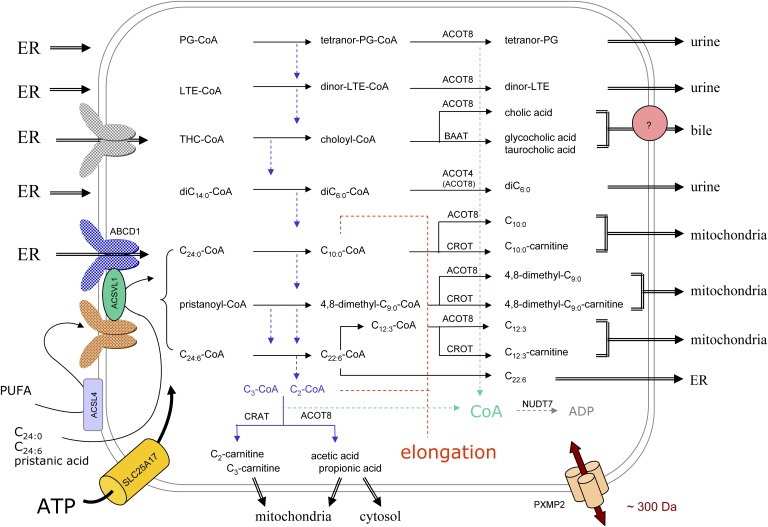
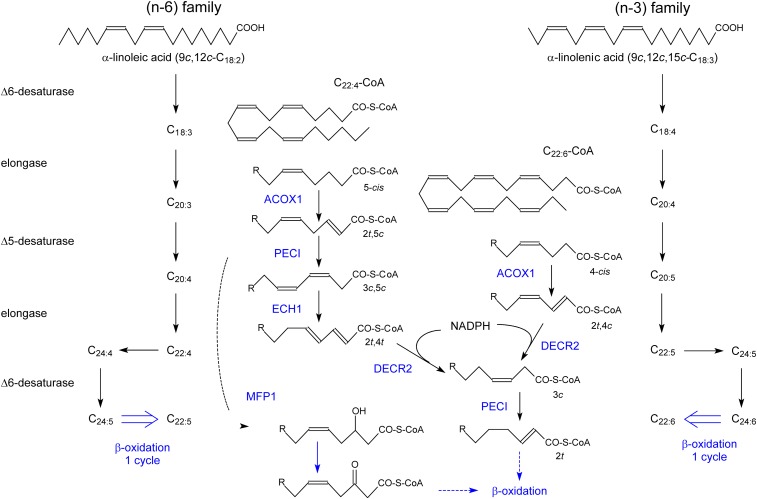
In humans, peroxisomes harbor a complex set of enzymes acting on various lipophilic carboxylic acids, organized in two basic pathways, alpha-oxidation and beta-oxidation; the latter pathway can also handle omega-oxidized compounds. Some oxidation products are crucial to human health (primary bile acids and polyunsaturated FAs), whereas other substrates have to be degraded in order to avoid neuropathology at a later age (very long-chain FAs and xenobiotic phytanic acid and pristanic acid). Whereas total absence of peroxisomes is lethal, single peroxisomal protein deficiencies can present with a mild or severe phenotype and are more informative to understand the pathogenic factors. The currently known single protein deficiencies equal about one-fourth of the number of proteins involved in peroxisomal FA metabolism. The biochemical properties of these proteins are highlighted, followed by an overview of the known diseases.
Figures
References
-
- Goldfischer S., Moore C. L., Johnson A. B., Spiro A. J., Valsamis M. P., Wisniewski H. K., Ritch R. H., Norton W. T., Rapin I., Gartner L. M. 1973. Peroxisomal and mitochondrial defects in the cerebro-hepato-renal syndrome. Science. 182: 62–64. - PubMed
-
- Kikuchi M., Hatano N., Yokota S., Shimozawa N., Imanaka T., Taniguchi H. 2004. Proteomic analysis of rat liver peroxisome: presence of peroxisome-specific isozyme of Lon protease. J. Biol. Chem. 279: 421–428. - PubMed
-
- Mi J., Kirchner E., Cristobal S. 2007. Quantitative proteomic comparison of mouse peroxisomes from liver and kidney. Proteomics. 7: 1916–1928. - PubMed
-
- Wiese S., Gronemeyer T., Ofman R., Kunze M., Grou C. P., Almeida J. A., Eisenacher M., Stephan C., Hayen H., Schollenberger L., et al. 2007. Proteomics characterization of mouse kidney peroxisomes by tandem mass spectrometry and protein correlation profiling. Mol. Cell. Proteomics. 6: 2045–2057. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
