

Hyperoxic stimulation of synchronous orthodromic activity and induction of neural plasticity does not require changes in excitatory synaptic transmission
- PMID: 20558752
- PMCID: PMC2944636
- DOI: 10.1152/japplphysiol.91430.2008
Hyperoxic stimulation of synchronous orthodromic activity and induction of neural plasticity does not require changes in excitatory synaptic transmission
Abstract
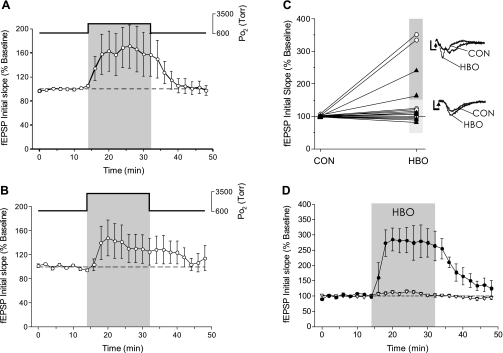
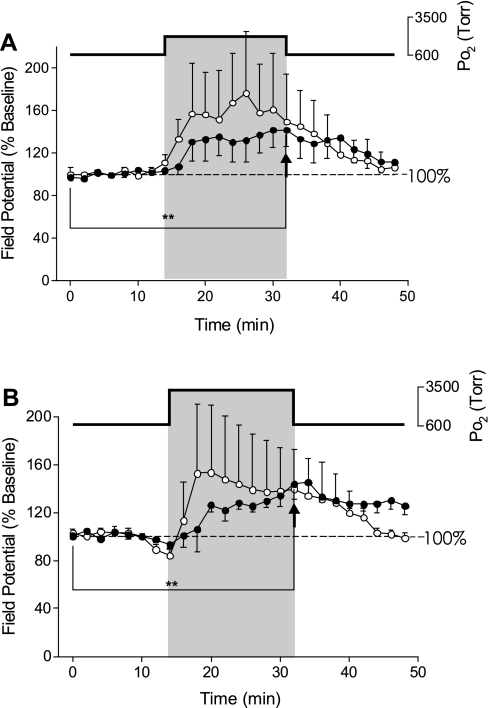
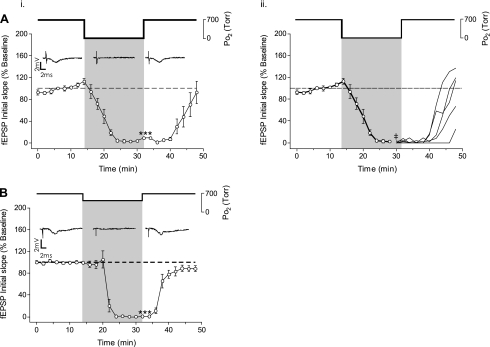
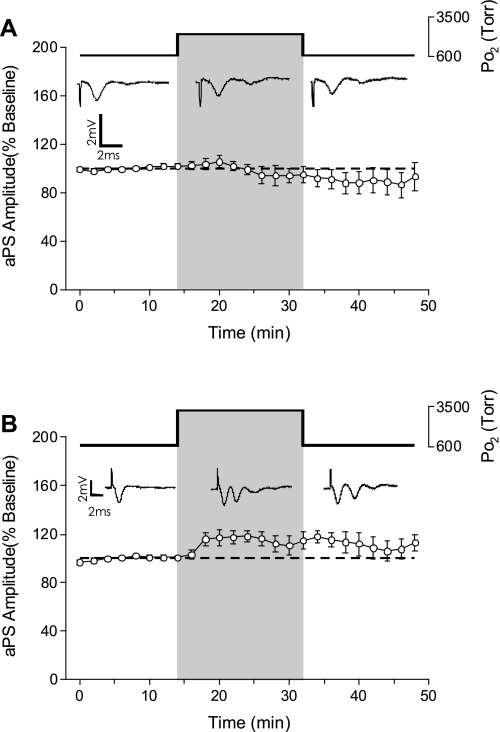
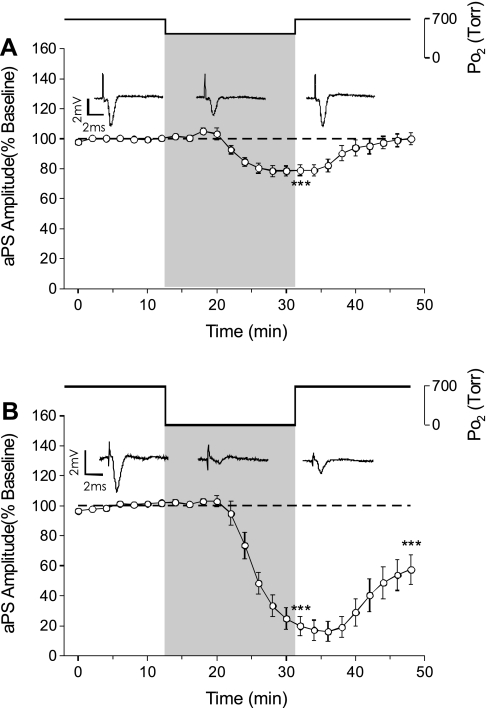
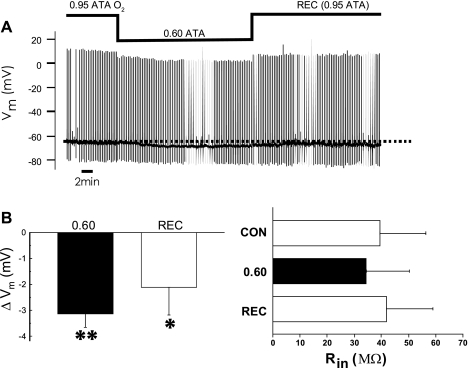
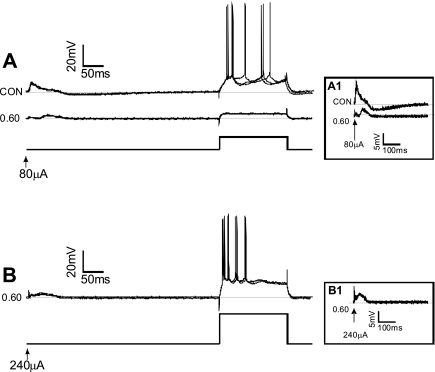
The first study, described in the companion article, reports that acute exposure of rat hippocampal slices to either hyperbaric oxygen (HBO: 2.84 and 4.54 atmospheres absolute, ATA) or normobaric reoxygenation (NBOreox; i.e., normobaric hyperoxia: 0.6 or 0.0→0.95 ATA) stimulates synchronous orthodromic activity in CA1 neurons, which includes activation of O2-induced potentiation (OxIP) and, in some cases, hyperexcitability (secondary population spikes, sPS). In this second study we tested the hypothesis that HBO and NBOreox increase orthodromic activity of CA1 neurons (oPS, orthodromic population spike) and OxIP via a combination of both increased excitatory synaptic transmission (field excitatory postsynaptic potential, fEPSP) and intrinsic excitability (antidromic population spike, aPS). HBO and NBOreox increased the oPS but rarely increased or potentiated the fEPSP. HBO exposure produced epileptiform antidromic activity, which was abolished during inhibition of fast GABAergic and glutamatergic synaptic transmission. Decreasing O2 from 0.95 ATA (control) to 0.6 ATA (intermediate O2) or 0.0 ATA (hypoxia) reversibly abolished the fEPSP, and reoxygenation rarely induced potentiation of the fEPSP or aPS. Intracellular recordings and antidromic field potential recordings, however, revealed that synaptic transmission and neuronal excitability were preserved, albeit at lower levels, in 0.60 ATA O2. Together, these data indicate that 1) the changes in excitatory postsynaptic activity are not required for stimulation of the oPS during and HBO/NBOreox or for activation of OxIP, suggesting the latter is a form of intrinsic plasticity; 2) HBO disinhibits spontaneous synaptic transmission to induce epileptiform activity; and 3) although synchronous synaptic activation of the CA1 neuronal population requires hyperoxia (i.e., 0.95 ATA O2), synaptic activation of individual CA1 neurons does not.
Figures



References
-
- Bitterman N, Halpern P. The effect of flumazenil on CNS oxygen toxicity in the rat. Methods Find Exp Clin Pharmacol 17: 169–174, 1995 - PubMed
-
- Brahman B, Forman RE, Stewart EE, Nicholson C, Rice ME. Ascorbate inhibits edema in brain slices. J Neurochem 74: 1263–1270, 2000 - PubMed
-
- D'Agostino DP, Putnam RW, Dean JB. Superoxide (•O2−) production in CA1 neurons of rat hippocampal slices exposed to graded levels of oxygen. J Neurophysiol 98: 1030–1041, 2007 - PubMed
-
- Daoudal G, Dubane D. Long-term plasticity of intrinsic excitability: learning rules and memory. Learn Mem 10: 456–465, 2003 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous