

Hyperbaric hyperoxia and normobaric reoxygenation increase excitability and activate oxygen-induced potentiation in CA1 hippocampal neurons
- PMID: 20558753
- PMCID: PMC2944633
- DOI: 10.1152/japplphysiol.91429.2008
Hyperbaric hyperoxia and normobaric reoxygenation increase excitability and activate oxygen-induced potentiation in CA1 hippocampal neurons
Abstract
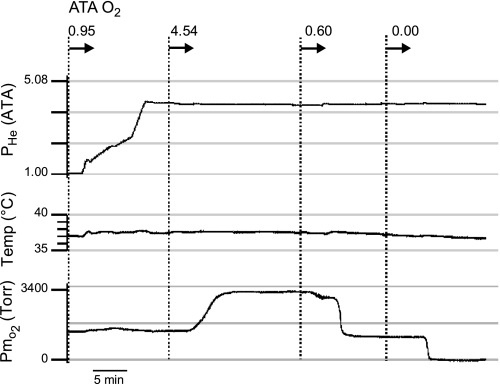
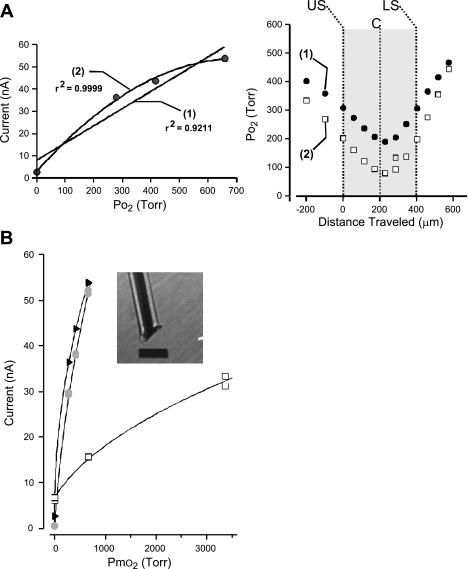
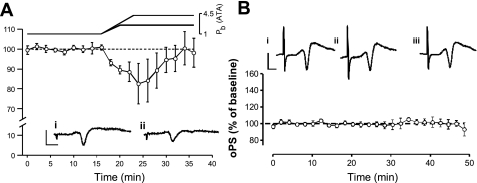
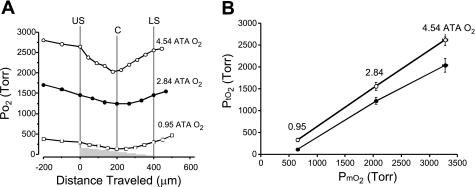
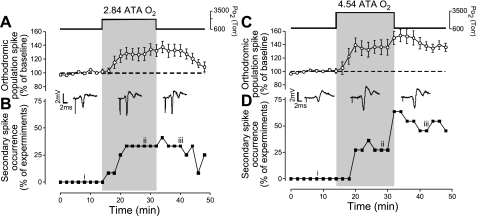
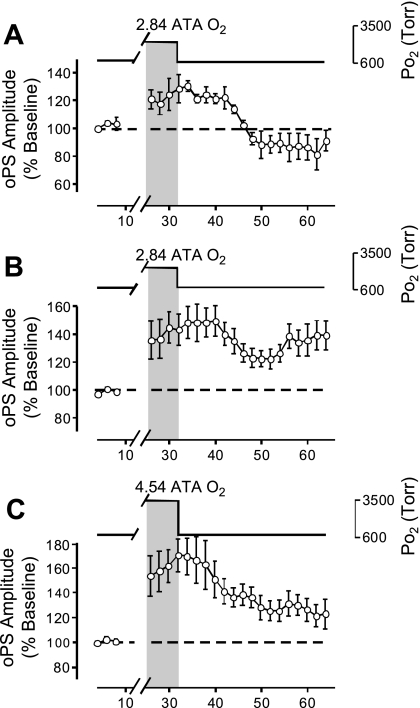
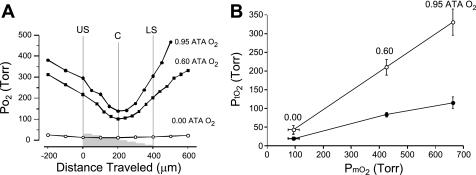
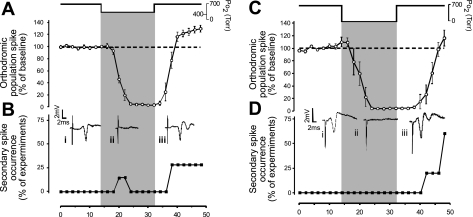
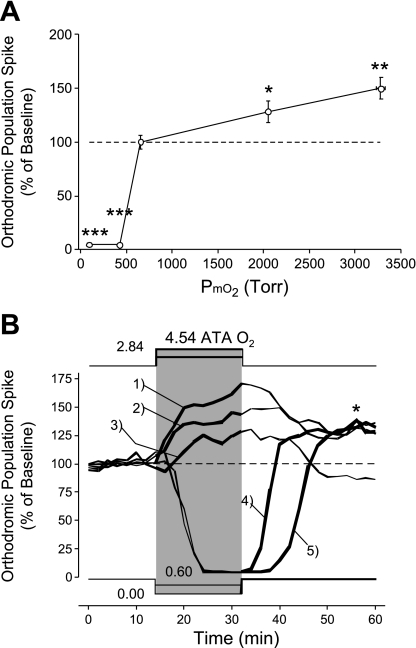
Breathing hyperbaric oxygen (HBO) is common practice in hyperbaric and diving medicine. The benefits of breathing HBO, however, are limited by the risk of central nervous system O2 toxicity, which presents as seizures. We tested the hypothesis that excitability increases in CA1 neurons of the rat hippocampal slice (400 microm) over a continuum of hyperoxia that spans normobaric and hyperbaric pressures. Amplitude changes of the orthodromic population spike were used to assess neuronal O2 sensitivity before, during, and following exposure to 0, 0.6, 0.95 (control), 2.84, and 4.54 atmospheres absolute (ATA) O2. Polarographic O2 electrodes were used to measure tissue slice PO2 (PtO2). In 0.95 ATA O2, core PtO2 at 200 microm deep was 115±16 Torr (mean±SE). Increasing O2 to 2.84 and 4.54 ATA increased core PtO2 to 1,222±77 and 2,037±157 Torr, respectively. HBO increased the orthodromic population spike amplitude and usually induced hyperexcitability (i.e., secondary population spikes) and, in addition, a long-lasting potentiation of the orthodromic population spike that we have termed "oxygen-induced potentiation" (OxIP). Exposure to 0.60 ATA O2 and hypoxia (0.00 ATA) decreased core PtO2 to 84±6 and 20±4 Torr, respectively, and abolished the orthodromic response. Reoxygenation from 0.0 or 0.6 ATA O2, however, usually produced a response similar to that of HBO: hyperexcitability and activation of OxIP. We conclude that CA1 neurons exhibit increased excitability and neural plasticity over a broad range of PtO2, which can be activated by a single, hyperoxic stimulus. We postulate that transient acute hyperoxia stimulus, whether caused by breathing HBO or reoxygenation following hypoxia (e.g., disordered breathing), is a powerful stimulant for orthodromic activity and neural plasticity in the CA1 hippocampus.
Figures
References
-
- Aitken PG, Breese GR, Dudek FF, Edwards F, Espanol MT, Larkman PM, Lipton P, Newman GC, Nowak TSJ, Panizzon KL. Preparative methods for brain slices: a discussion. J Neurosci Methods 59: 139–149, 1995 - PubMed
-
- Alger BE, Dhanjal SS, Dingledine R, Garthwaite J, Henderson G, King GL, Lipton P, North A, Schwartzkronin PA, Sears TA, Segal M, Whittingham TS, Williams J. Appendix, brain slice methods. In: Brain Slices, edited by Dingledine R. New York: Plenum, 1984, p. 381–437
-
- Arieli R, Gutterman A. Recovery time constant in central nervous system O2 toxicity in the rat. Eur J Appl Physiol 75: 182–187, 1997 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous