

Nitro-oleic acid inhibits angiotensin II-induced hypertension
- PMID: 20558825
- PMCID: PMC2937264
- DOI: 10.1161/CIRCRESAHA.110.218404
Nitro-oleic acid inhibits angiotensin II-induced hypertension
Abstract
Rationale: Nitro-oleic acid (OA-NO(2)) is a bioactive, nitric-oxide derived fatty acid with physiologically relevant vasculoprotective properties in vivo. OA-NO(2) exerts cell signaling actions as a result of its strong electrophilic nature and mediates pleiotropic cell responses in the vasculature.
Objective: The present study sought to investigate the protective role of OA-NO(2) in angiotensin (Ang) II-induced hypertension.
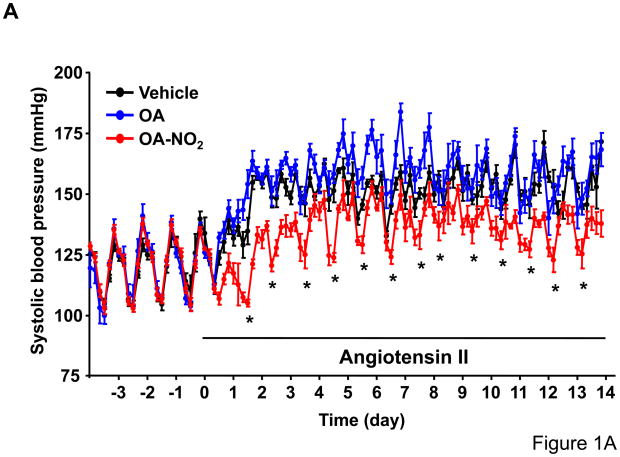
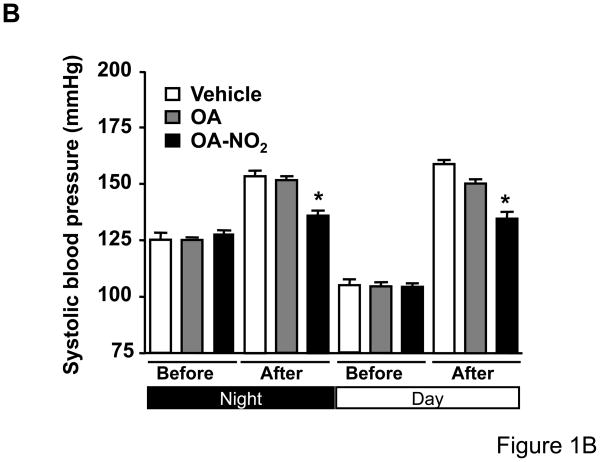
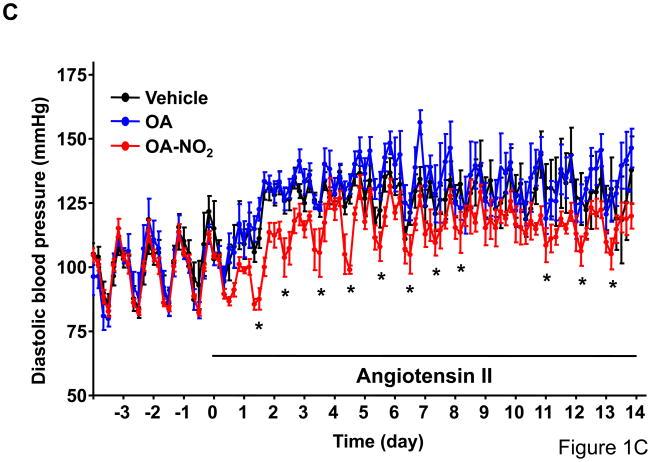
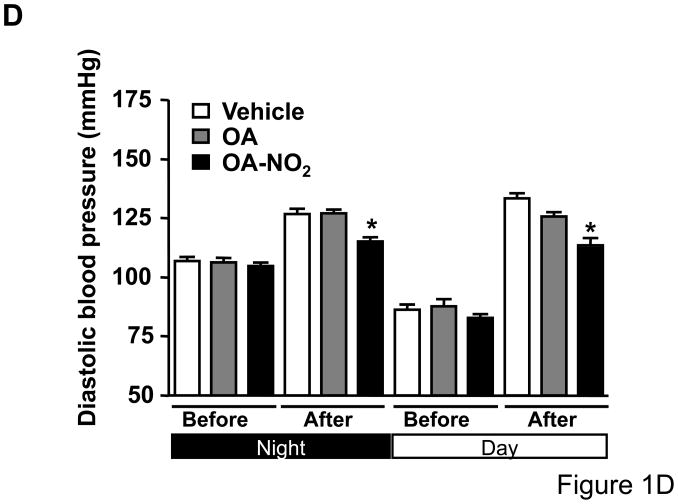
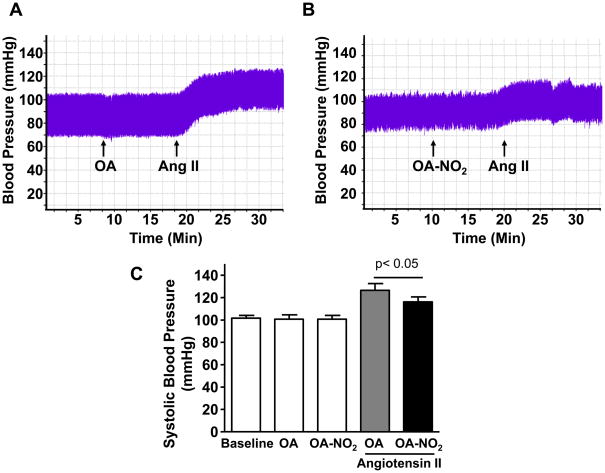
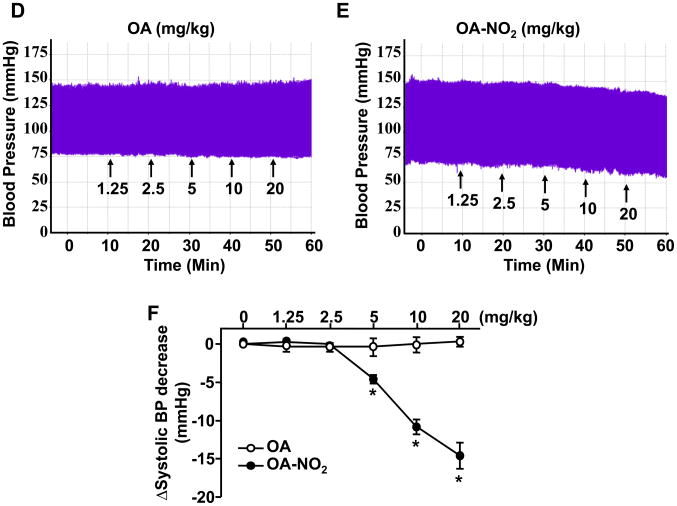
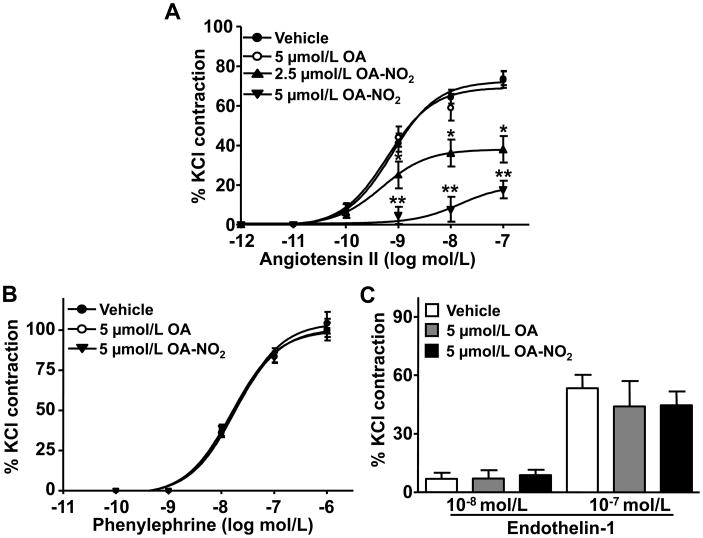
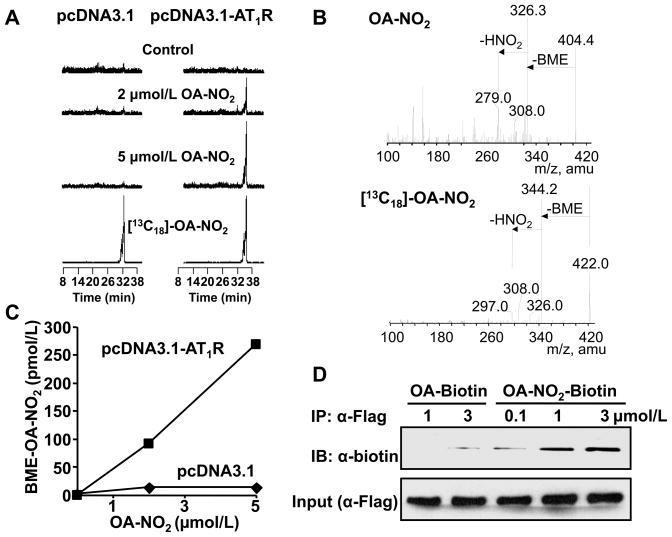
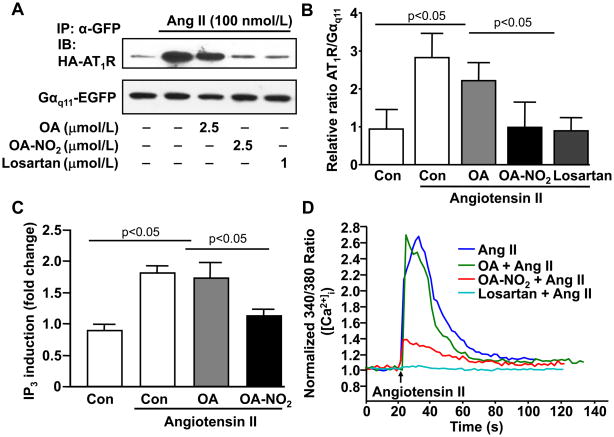
Methods and results: We show that systemic administration of OA-NO(2) results in a sustained reduction of Ang II-induced hypertension in mice and exerts a significant blood pressure lowering effect on preexisting hypertension established by Ang II infusion. OA-NO(2) significantly inhibits Ang II contractile response as compared to oleic acid (OA) in mesenteric vessels. The improved vasoconstriction is specific for the Ang II type 1 receptor (AT(1)R)-mediated signaling because vascular contraction by other G-protein-coupled receptors is not altered in response to OA-NO(2) treatment. From the mechanistic viewpoint, OA-NO(2) lowers Ang II-induced hypertension independently of peroxisome proliferation-activated receptor (PPAR)gamma activation. Rather, OA-NO(2), but not OA, specifically binds to the AT(1)R, reduces heterotrimeric G-protein coupling, and inhibits IP(3) (inositol-1,4,5-trisphosphate) and calcium mobilization, without inhibiting Ang II binding to the receptor.
Conclusions: These results demonstrate that OA-NO(2) diminishes the pressor response to Ang II and inhibits AT(1)R-dependent vasoconstriction, revealing OA-NO(2) as a novel antagonist of Ang II-induced hypertension.
Figures


Similar articles
-
Oleic Acid Attenuates Ang II (Angiotensin II)-Induced Cardiac Remodeling by Inhibiting FGF23 (Fibroblast Growth Factor 23) Expression in Mice.Hypertension. 2020 Mar;75(3):680-692. doi: 10.1161/HYPERTENSIONAHA.119.14167. Epub 2020 Jan 13. Hypertension. 2020. PMID: 31928110
-
Protective actions of estrogen on angiotensin II-induced hypertension: role of central nitric oxide.Am J Physiol Heart Circ Physiol. 2009 Nov;297(5):H1638-46. doi: 10.1152/ajpheart.00502.2009. Epub 2009 Sep 4. Am J Physiol Heart Circ Physiol. 2009. PMID: 19734362 Free PMC article.
-
Suppression of Vascular Macrophage Activation by Nitro-Oleic Acid and its Implication for Abdominal Aortic Aneurysm Therapy.Cardiovasc Drugs Ther. 2021 Oct;35(5):939-951. doi: 10.1007/s10557-020-07031-8. Epub 2020 Jul 15. Cardiovasc Drugs Ther. 2021. PMID: 32671602 Free PMC article.
-
AT2 Receptor Activation Prevents Sodium Retention and Reduces Blood Pressure in Angiotensin II-Dependent Hypertension.Circ Res. 2016 Aug 5;119(4):532-43. doi: 10.1161/CIRCRESAHA.116.308384. Epub 2016 Jun 20. Circ Res. 2016. PMID: 27323774 Free PMC article.
-
[Angiotensin type 2 receptor-dependent vasodilation].Yakugaku Zasshi. 2007 Sep;127(9):1357-67. doi: 10.1248/yakushi.127.1357. Yakugaku Zasshi. 2007. PMID: 17827918 Review. Japanese.
Cited by
-
Redox Regulation of Soluble Epoxide Hydrolase-Implications for Cardiovascular Health and Disease.Cells. 2022 Jun 15;11(12):1932. doi: 10.3390/cells11121932. Cells. 2022. PMID: 35741062 Free PMC article. Review.
-
A novel nitroalkene-α-tocopherol analogue inhibits inflammation and ameliorates atherosclerosis in Apo E knockout mice.Br J Pharmacol. 2019 Mar;176(6):757-772. doi: 10.1111/bph.14561. Epub 2019 Feb 3. Br J Pharmacol. 2019. PMID: 30588602 Free PMC article.
-
Post-Translational Modification of Proteins Mediated by Nitro-Fatty Acids in Plants: Nitroalkylation.Plants (Basel). 2019 Mar 29;8(4):82. doi: 10.3390/plants8040082. Plants (Basel). 2019. PMID: 30934982 Free PMC article. Review.
-
Nitro-Fatty Acid Logistics: Formation, Biodistribution, Signaling, and Pharmacology.Trends Endocrinol Metab. 2019 Aug;30(8):505-519. doi: 10.1016/j.tem.2019.04.009. Epub 2019 Jun 10. Trends Endocrinol Metab. 2019. PMID: 31196614 Free PMC article. Review.
-
The role of nutrition and nutraceutical supplements in the treatment of hypertension.World J Cardiol. 2014 Feb 26;6(2):38-66. doi: 10.4330/wjc.v6.i2.38. World J Cardiol. 2014. PMID: 24575172 Free PMC article. Review.
References
-
- Lundberg JO, Gladwin MT, Ahluwalia A, Benjamin N, Bryan NS, Butler A, Cabrales P, Fago A, Feelisch M, Ford PC, Freeman BA, Frenneaux M, Friedman J, Kelm M, Kevil CG, Kim-Shapiro DB, Kozlov AV, Lancaster JR, Jr, Lefer DJ, McColl K, McCurry K, Patel RP, Petersson J, Rassaf T, Reutov VP, Richter-Addo GB, Schechter A, Shiva S, Tsuchiya K, van Faassen EE, Webb AJ, Zuckerbraun BS, Zweier JL, Weitzberg E. Nitrate and nitrite in biology, nutrition and therapeutics. Nat Chem Biol. 2009;5:865–869. - PMC - PubMed
-
- Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO, 3rd, Gladwin MT. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498–1505. - PubMed
-
- Jansson EA, Huang L, Malkey R, Govoni M, Nihlen C, Olsson A, Stensdotter M, Petersson J, Holm L, Weitzberg E, Lundberg JO. A mammalian functional nitrate reductase that regulates nitrite and nitric oxide homeostasis. Nat Chem Biol. 2008;4:411–417. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 EB008999/EB/NIBIB NIH HHS/United States
- R01 HL089544/HL/NHLBI NIH HHS/United States
- HL089544/HL/NHLBI NIH HHS/United States
- HL64937/HL/NHLBI NIH HHS/United States
- K99 HL095769/HL/NHLBI NIH HHS/United States
- R01 HL105114/HL/NHLBI NIH HHS/United States
- R01 HL088391/HL/NHLBI NIH HHS/United States
- R01 CA116592/CA/NCI NIH HHS/United States
- R01 HL058115/HL/NHLBI NIH HHS/United States
- R01 HL068878/HL/NHLBI NIH HHS/United States
- R37 HL058115/HL/NHLBI NIH HHS/United States
- CA116592/CA/NCI NIH HHS/United States
- HL68878/HL/NHLBI NIH HHS/United States
- R01 HL064937/HL/NHLBI NIH HHS/United States
- R01 HL075397/HL/NHLBI NIH HHS/United States
- HL58115/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
