

Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors
- PMID: 20559783
- PMCID: PMC2900478
- DOI: 10.1007/s12265-010-9174-x
Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors
Abstract
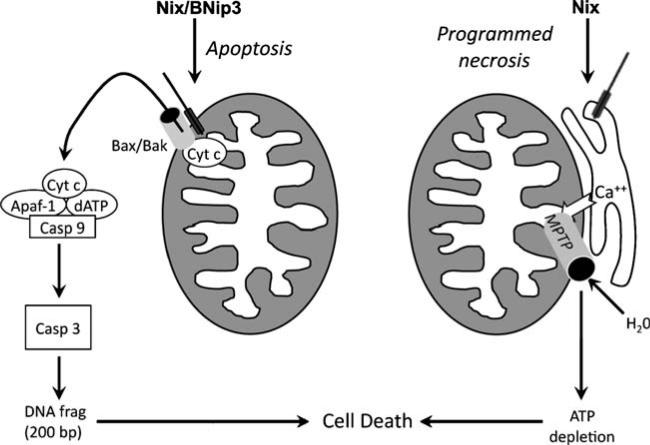
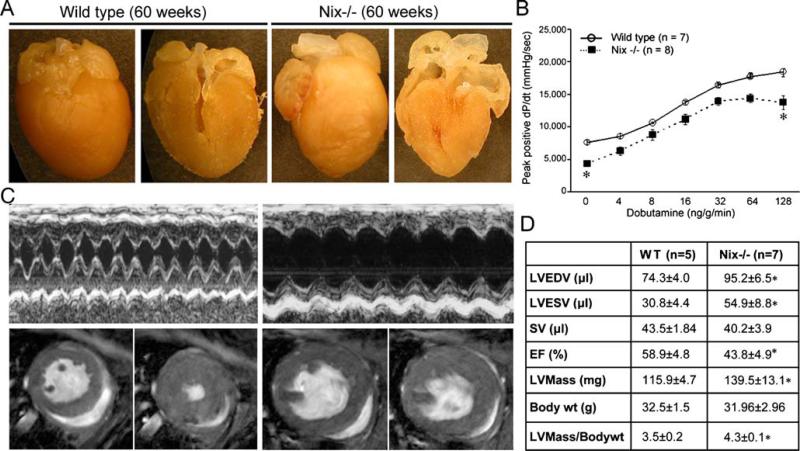
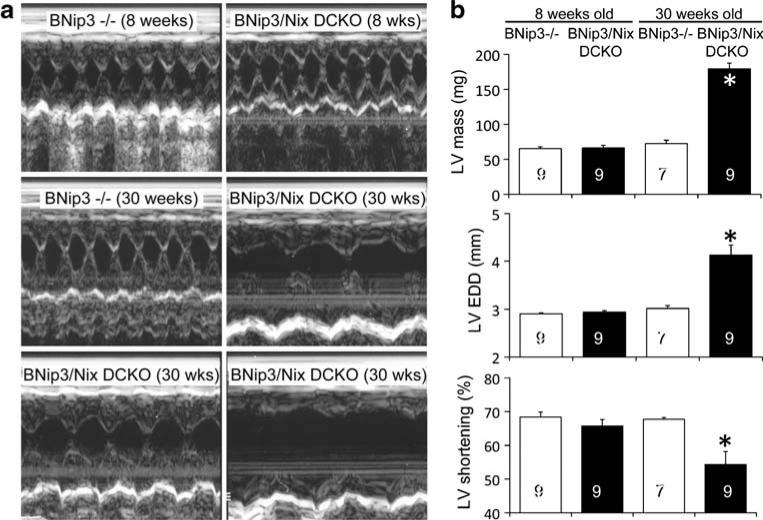
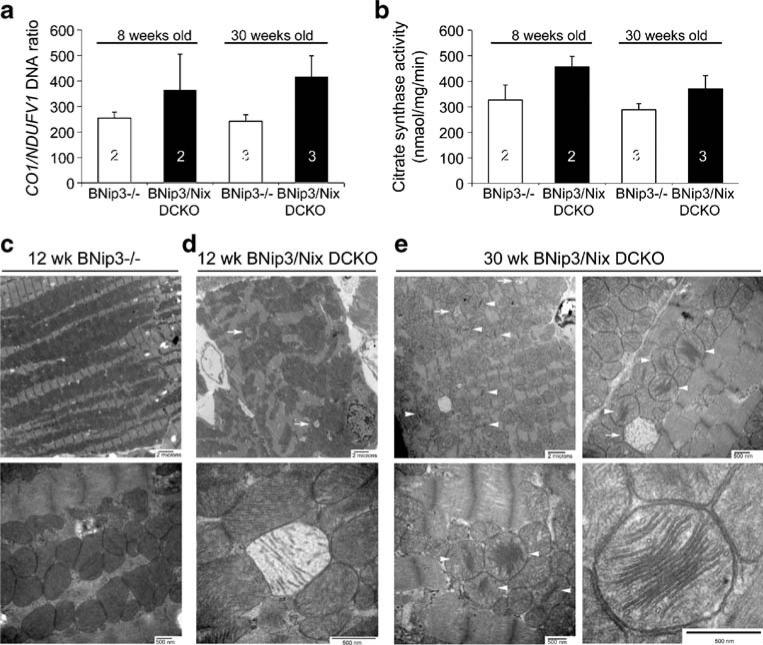
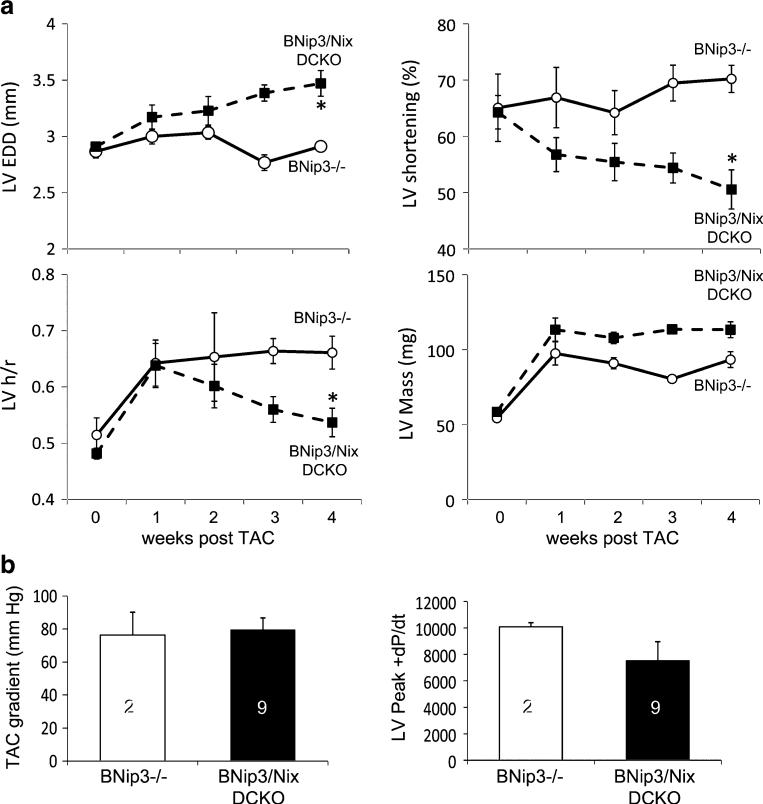
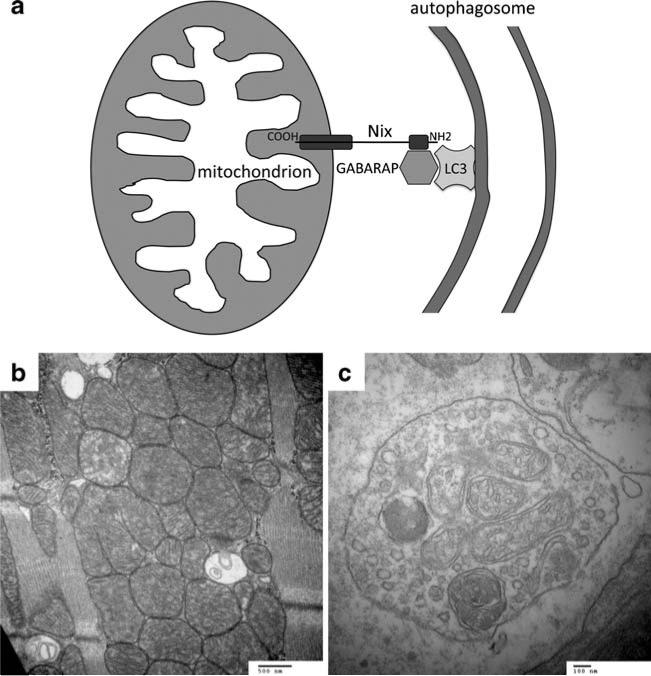
Programmed cardiac myocyte death via the intrinsic, or mitochondrial, pathway is a mechanism of pathological ventricular remodeling after myocardial infarction and during chronic pressure overload hypertrophy. Transcriptional upregulation of the closely related proapoptotic Bcl2 family members BNip3 in ischemic myocardium and Nix in hypertrophied myocardium suggested a molecular mechanism by which programmed cell death can be initiated by cardiac stress and lead to dilated cardiomyopathy. Studies using transgenic and gene knockout mice subsequently demonstrated that expression of BNip3 and Nix is both sufficient for cardiomyopathy development and necessary for cardiac remodeling after reversible coronary occlusion and transverse aortic banding, respectively. Here, these data are reviewed in the context of recent findings showing that Nix not only stimulates cardiomyocyte apoptosis but also induces mitochondrial autophagy (mitophagy) and indirectly activates the mitochondrial permeability transition pore, causing cell necrosis. New findings are presented suggesting that Nix and BNip3 have an essential function, "mitochondrial pruning," that restrains mitochondrial proliferation in cardiomyocytes and without which an age-dependent mitochondrial cardiomyopathy develops.
Figures
References
-
- Diwan A, Dorn GW., II Decompensation of cardiac hypertrophy: Cellular mechanisms and novel therapeutic targets. Physiology (Bethesda) 2007;22:56–64. - PubMed
-
- Berry JJ, Hoffman JM, Steenbergen C, Baker JA, Floyd C, Van Trigt P, et al. Human pathologic correlation with PET in ischemic and nonischemic cardiomyopathy. Journal of Nuclear Medicine. 1993;34:39–47. - PubMed
-
- Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, et al. Apoptosis in the failing human heart. Journal of Nuclear Medicine. 1997;336:1131–1141. - PubMed
-
- Francis GS. Changing the remodeling process in heart failure: Basic mechanisms and laboratory results. Current Opinion in Cardiology. 1998;13:156–161. - PubMed
-
- Olivetti G, Capasso JM, Sonnenblick EH, Anversa P. Side-to-side slippage of myocytes participates in ventricular wall remodeling acutely after myocardial infarction in rats. Circulation Research. 1990;67:23–34. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
