

Loss-of-function mutations in HPSE2 cause the autosomal recessive urofacial syndrome
- PMID: 20560209
- PMCID: PMC3032074
- DOI: 10.1016/j.ajhg.2010.04.016
Loss-of-function mutations in HPSE2 cause the autosomal recessive urofacial syndrome
Erratum in
- Am J Hum Genet. 2010 Jul 9;87(1):161. Fisher, Richard B [added]
Abstract
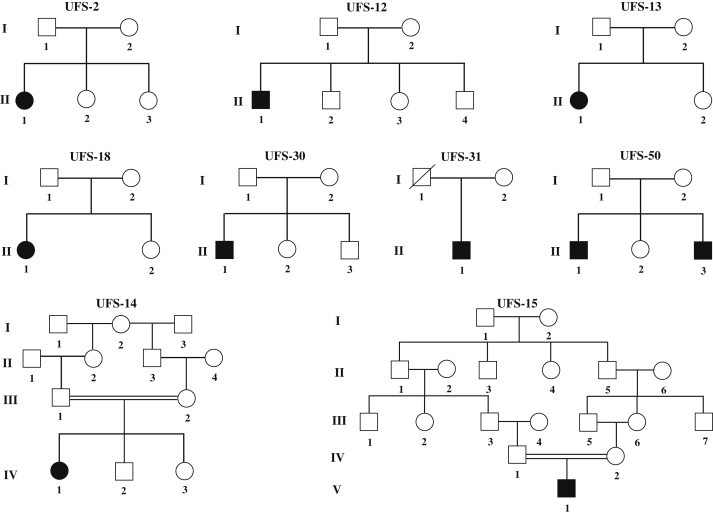
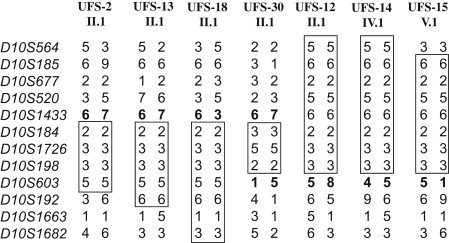
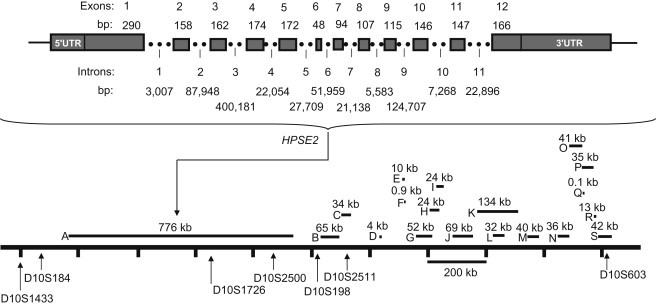
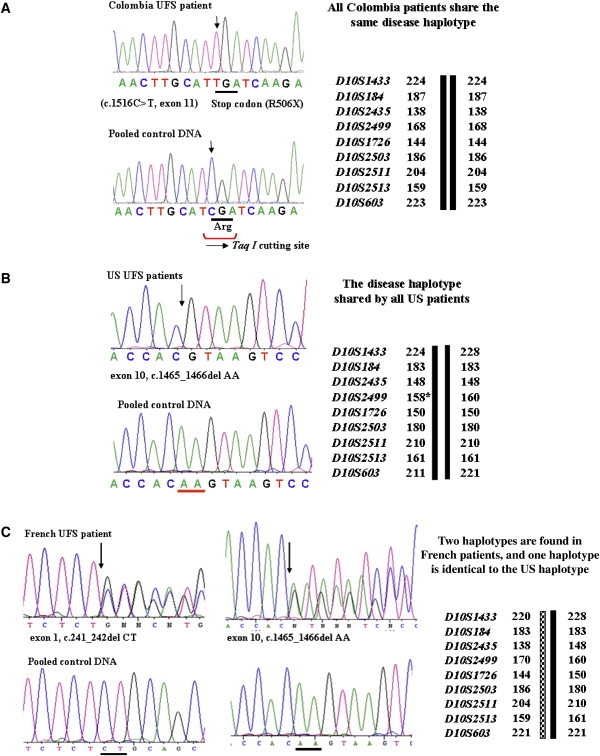
Previously, we localized the defective gene for the urofacial syndrome (UFS) to a region on chromosome 10q24 by homozygosity mapping. We now report evidence that Heparanse 2 (HPSE2) is the culprit gene for the syndrome. Mutations with a loss of function in the Heparanase 2 (HPSE2) gene were identified in all UFS patients originating from Colombia, the United States, and France. HPSE2 encodes a 592 aa protein that contains a domain showing sequence homology to the glycosyl hydrolase motif in the heparanase (HPSE) gene, but its exact biological function has not yet been characterized. Complete loss of HPSE2 function in UFS patients suggests that HPSE2 may be important for the synergic action of muscles implicated in facial expression and urine voiding.
Figures
References
-
- Ochoa B. The urofacial (Ochoa) syndrome revisited. J. Urol. 1992;148:580–583. - PubMed
-
- Ochoa B., Gorlin R.J. Urofacial (ochoa) syndrome. Am. J. Med. Genet. 1987;27:661–667. - PubMed
-
- Ochoa B. Can a congenital dysfunctional bladder be diagnosed from a smile? The Ochoa syndrome updated. Pediatr. Nephrol. 2004;19:6–12. - PubMed
-
- Wang C.Y., Huang Y.Q., Shi J.D., Marron M.P., Ruan Q.G., Hawkins-Lee B., Ochoa B., She J.X. Genetic homogeneity, high-resolution mapping, and mutation analysis of the urofacial (Ochoa) syndrome and exclusion of the glutamate oxaloacetate transaminase gene (GOT1) in the critical region as the disease gene. Am. J. Med. Genet. 1999;84:454–459. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases