

Mutations in HPSE2 cause urofacial syndrome
- PMID: 20560210
- PMCID: PMC3032078
- DOI: 10.1016/j.ajhg.2010.05.006
Mutations in HPSE2 cause urofacial syndrome
Erratum in
- Am J Hum Genet. 2010 Aug 13;87(2):309
Abstract
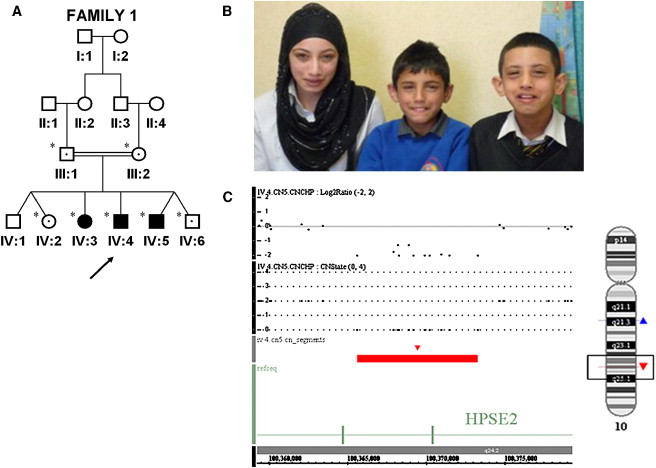
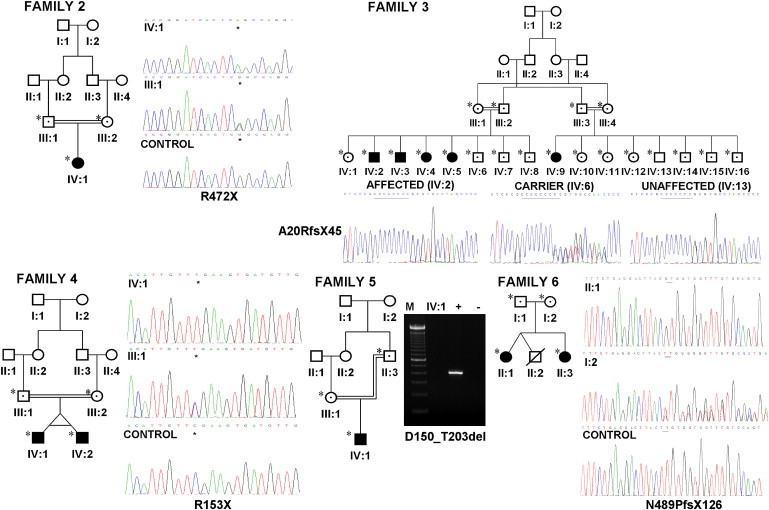
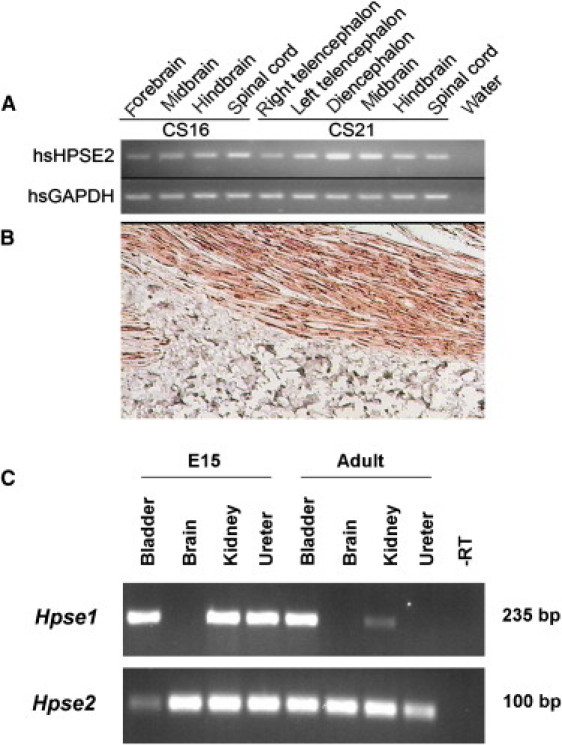
Urinary voiding dysfunction in childhood, manifesting as incontinence, dysuria, and urinary frequency, is a common condition. Urofacial syndrome (UFS) is a rare autosomal recessive disease characterized by facial grimacing when attempting to smile and failure of the urinary bladder to void completely despite a lack of anatomical bladder outflow obstruction or overt neurological damage. UFS individuals often have reflux of infected urine from the bladder to the upper renal tract, with a risk of kidney damage and renal failure. Whole-genome SNP mapping in one affected individual defined an autozygous region of 16 Mb on chromosome 10q23-q24, within which a 10 kb deletion encompassing exons 8 and 9 of HPSE2 was identified. Homozygous exonic deletions, nonsense mutations, and frameshift mutations in five further unrelated families confirmed HPSE2 as the causative gene for UFS. Mutations were not identified in four additional UFS patients, indicating genetic heterogeneity. We show that HPSE2 is expressed in the fetal and adult central nervous system, where it might be implicated in controlling facial expression and urinary voiding, and also in bladder smooth muscle, consistent with a role in renal tract morphology and function. Our findings have broader implications for understanding the genetic basis of lower renal tract malformations and voiding dysfunction.
Figures
References
-
- Hoebeke P., van der Walle J. Voiding dysfunction, recurrent urinary infection, constipation, and vesicoureteric reflux: A common disease complex. Dialog. Pediatr. Urol. 2002;25:2–3.
-
- Ochoa B., Gorlin R.J. Urofacial (ochoa) syndrome. Am. J. Med. Genet. 1987;27:661–667. - PubMed
-
- Ochoa B. The urofacial (Ochoa) syndrome revisited. J. Urol. 1992;148:580–583. - PubMed
-
- Wang C.Y., Huang Y.Q., Shi J.D., Marron M.P., Ruan Q.G., Hawkins-Lee B., Ochoa B., She J.X. Genetic homogeneity, high-resolution mapping, and mutation analysis of the urofacial (Ochoa) syndrome and exclusion of the glutamate oxaloacetate transaminase gene (GOT1) in the critical region as the disease gene. Am. J. Med. Genet. 1999;84:454–459. - PubMed
-
- Wang C.Y., Davoodi-Semiromi A., Shi J.D., Yang P., Huang Y.Q., Agundez J.A., Moran J.M., Ochoa B., Hawkins-Lee B., She J.X. High resolution mapping and mutation analyses of candidate genes in the urofacial syndrome (UFS) critical region. Am. J. Med. Genet. A. 2003;119A:9–14. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous